

Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) is a renal tubular disorder characterised by excessive urinary excretion of magnesium and calcium and progressive deterioration in kidney function, with progression to chronic kidney disease attributed to calcium deposits in the renal parenchyma.1 It is transmitted with autosomal recessive inheritance, associated with the mutation in the CLDN16 and 19 genes, which encode claudin-16 and 19, involved in the paracellular transport of calcium and magnesium through tight junctions, intercellular junction zones of the apical membrane in the thick segment of the ascending limb of the loop of Henle.2–5
The signs and symptoms include those directly related to renal tubular disorders, such as polyuria, polydipsia, lithiasis, nephrocalcinosis and recurrent infections. Other associated symptoms have been reported, such as neuromuscular and eye disorders.3–6
We present the case of a 29-year-old male with a clinical history since childhood of moderate sensorineural hearing loss under follow-up by the Ear, Nose and Throat service; in addition he had t horizontal nystagmus, myopia and astigmatism under follow-up by Ophthalmology, and paranoid disorder with poor treatment adherence being followed by Psychiatry.
Evaluation of his family history revealed that he had two healthy brothers, and in addition there was collateral consanguinity, as his parents were cousins.
In March 2020, he was referred to the Nephrology clinic due to progressive deterioration in kidney function, with creatinine 2.03 mg/dl and estimated glomerular filtration rate (eGFR) 41 ml/min, and sterile pyuria; in May 2017, he had a eGFR of 52 ml/min (Cr 1.76 mg/dl). He reported being clinically asymptomatic, although he was maintaining polyuria of about 2-3 l (V/GFR = 1.88) and habitual polydipsia, not perceived by the patient to be excessive.
Ultrasound showed kidneys with a chronic appearance, reduced in size, and hyperechoic material in the medulla consistent with nephrocalcinosis (Fig. 1).

The patient was given an appointment to return to the Nephrology clinic for further tests. However, he went to Emergency room with a three-week history of a "tingling" sensation and cramps, that was worsening in the previous days, and he was admitted to Nephrology for symptomatic treatment and to complete the evaluation.
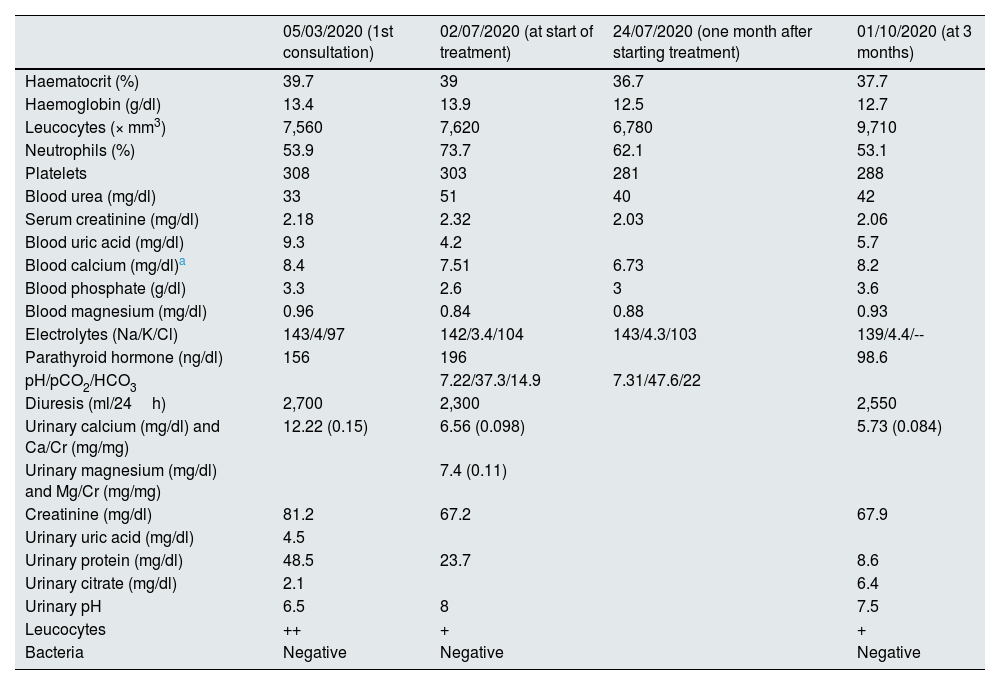
Lab tests showed deterioration in renal function, with hypocalcaemia (7.7 mg/dl), hypomagnesaemia (0.96 mg/dl), normochloraemic metabolic acidosis, increased anion gap, hyperuricaemia and hyperparathyroidism (Table 1).
Changes in biochemical data in blood and urine from the first Nephrology consultation, at diagnosis, at one month and at three months of treatment.
| 05/03/2020 (1st consultation) | 02/07/2020 (at start of treatment) | 24/07/2020 (one month after starting treatment) | 01/10/2020 (at 3 months) | |
|---|---|---|---|---|
| Haematocrit (%) | 39.7 | 39 | 36.7 | 37.7 |
| Haemoglobin (g/dl) | 13.4 | 13.9 | 12.5 | 12.7 |
| Leucocytes (× mm3) | 7,560 | 7,620 | 6,780 | 9,710 |
| Neutrophils (%) | 53.9 | 73.7 | 62.1 | 53.1 |
| Platelets | 308 | 303 | 281 | 288 |
| Blood urea (mg/dl) | 33 | 51 | 40 | 42 |
| Serum creatinine (mg/dl) | 2.18 | 2.32 | 2.03 | 2.06 |
| Blood uric acid (mg/dl) | 9.3 | 4.2 | 5.7 | |
| Blood calcium (mg/dl)a | 8.4 | 7.51 | 6.73 | 8.2 |
| Blood phosphate (g/dl) | 3.3 | 2.6 | 3 | 3.6 |
| Blood magnesium (mg/dl) | 0.96 | 0.84 | 0.88 | 0.93 |
| Electrolytes (Na/K/Cl) | 143/4/97 | 142/3.4/104 | 143/4.3/103 | 139/4.4/-- |
| Parathyroid hormone (ng/dl) | 156 | 196 | 98.6 | |
| pH/pCO2/HCO3 | 7.22/37.3/14.9 | 7.31/47.6/22 | ||
| Diuresis (ml/24h) | 2,700 | 2,300 | 2,550 | |
| Urinary calcium (mg/dl) and Ca/Cr (mg/mg) | 12.22 (0.15) | 6.56 (0.098) | 5.73 (0.084) | |
| Urinary magnesium (mg/dl) and Mg/Cr (mg/mg) | 7.4 (0.11) | |||
| Creatinine (mg/dl) | 81.2 | 67.2 | 67.9 | |
| Urinary uric acid (mg/dl) | 4.5 | |||
| Urinary protein (mg/dl) | 48.5 | 23.7 | 8.6 | |
| Urinary citrate (mg/dl) | 2.1 | 6.4 | ||
| Urinary pH | 6.5 | 8 | 7.5 | |
| Leucocytes | ++ | + | + | |
| Bacteria | Negative | Negative | Negative |
The patient's urine had a pH of 8, with elevated magnesium excretion and decreased uric acid excretion. Urinary calcium excretion was within normal levels, although abnormally high in relation to blood levels (Table 1). Urinary oxalate excretion was normal.
The findings led to the diagnosis of a renal tubular disorder with excessive urinary loss of magnesium and calcium. Both the family history, with data of consanguinity, and the association of eye disorders since childhood were consistent with FHHNC.
For all these reasons, a genetic analysis was requested, which identified a homozygous change in the CLDN19 gene (c.59G>A (p.G20D), described in the literature as “Spanish/Hispanic or founder mutation". This causes the change of glycine for aspartic acid in position 20 of claudin-19, which alters the signal peptide and causes the mutant protein to be retained inside the cell, not being able to reach its location in the cell membrane.1,2
Treatment was prescribed with allopurinol, calcium carbonate, potassium citrate, sodium bicarbonate and magnesium lactate, with good clinical progress, although magnesium and calcium were difficult to control. At the follow-up visits in the subsequent months, no further deterioration in renal function was observed.
The association of eye disorders in FHHNC, such as nystagmus, myopia and coloboma, has been well defined and studied in the literature, and is closely related to the mutation of the CLDN19 gene (in up to 90% of patients), but not with the CLDN16 gene.5,6
However, it is unknown whether there might be an association between FHHNC and hearing impairments. Various studies have shown that hypomagnesaemia increases susceptibility to the development of sensorineural hearing loss. It is known that magnesium has an important neuroprotective and vasodilatory effect on hearing.7,8 The persistently low levels of magnesium in these patients with FHHNC could predispose them to suffer from sensorineural hearing loss or to suffer sudden deafness, as in our case reported here.
Mutations in other claudins have been related to hearing disorders, such as the mutation of the CLDN14 gene in autosomal recessive deafness DFNB29.9 Claudins 14 and 19 are found in the epithelium of the hair cells of the cochlea, and their expression in postnatal development has been described in murine models.10 However, to date it remains unknown whether mutations in the CLDN19 gene could cause hearing disorders, whether or not associated with FHHNC.
FundingThis study received no specific funding from public, private or non-profit organisations.
Conflicts of interestNone.



