

La hipofosfatemia ligada al cromosomaX (HLX) tiene una incidencia de 1/20.000 recién nacidos1. Está causada por mutaciones con pérdida de función del gen PHEX, que provocan un exceso del factor de crecimiento fibroblástico23 (FGF23) circulante, con pérdida renal de fosfato y menor síntesis de 1,25-dihidroxivitaminaD2. La hipofosfatemia crónica conduce a raquitismo y osteomalacia, retraso en el crecimiento, malformaciones de las extremidades inferiores, dolor y disminución de la función física, de la movilidad y de la calidad de vida1,3. La terapia de reemplazo convencional (sales de fosfato y 1,25-dihidroxivitaminaD)4 no normaliza la fosfatemia y puede inducir hipercalciuria, nefrocalcinosis e hiperparatiroidismo. Además, es difícil de implementar, en especial en niños, dados los efectos secundarios gastrointestinales y la necesidad de dosis frecuentes3-5. Burosumab (un anticuerpo monoclonal recombinante humano contra FGF23) mejora la reabsorción tubular renal del fosfato (RTP) con aumento de los niveles de fósforo y 1,25-dihidroxivitaminaD6.
Se realizó un estudio observacional, longitudinal y retrospectivo en un hospital de referencia de Bahía Blanca, Argentina, cuyo objetivo fue informar el resultado a largo plazo de los pacientes con raquitismo HLX confirmado genéticamente desde 1997 hasta 2022. Se registraron datos demográficos y clínicos al diagnóstico, estudios por imágenes y resultados de laboratorio iniciales y subsiguientes. Se realizaron controles cada 3 a 6meses y un examen odontológico anual. Se recolectó una muestra de mucosa yugal de un miembro de cada familia para un panel de secuenciación de nueva generación de 13 genes con diferentes patrones de herencia. También se obtuvieron datos relacionados con los tratamientos.
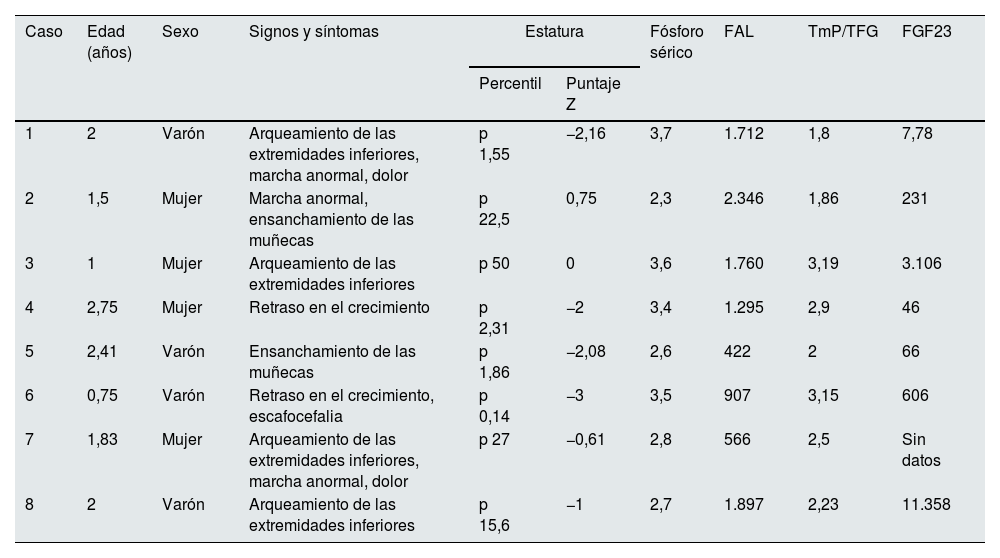
Se incluyeron ocho pacientes (cuatro varones) con una mediana de edad al diagnóstico de 1,9años (rango: 0,75-2,75), seguidos durante una mediana de 7,16años (rango: 2-24). Seis pacientes eran miembros de dos familias; los otros dos pacientes tenían variantes de novo que no habían sido descritas previamente (tabla 1). Las manifestaciones clínicas y los datos del laboratorio al diagnóstico se muestran en la tabla 2. La mitad tenían retraso del crecimiento; se observó estatura baja desproporcionada, malformaciones de las extremidades inferiores y marcha anormal en la mayoría de los pacientes. Todos presentaban limitaciones para realizar actividades diarias (saltar, correr, caminar), anomalías radiológicas severas (metáfisis en copa, epífisis ensanchadas e irregulares de los huesos largos), niveles bajos de fósforo, valores normales de calcio y parathormona, y niveles altos de fosfatasa alcalina. Cuatro pacientes tenían RTP <85% y todos los participantes se caracterizaban por reabsorción tubular máxima de fosfato por tasa de filtración glomerular (TmP/TFG) <3,8mg/dl. Los niveles de FGF23 eran variables (tabla 2).
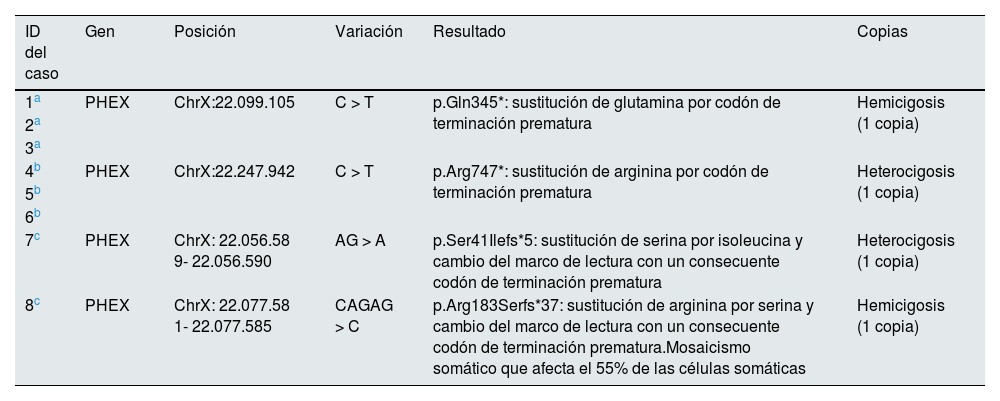
Mutación del gen PHEX
| ID del caso | Gen | Posición | Variación | Resultado | Copias |
|---|---|---|---|---|---|
| 1a | PHEX | ChrX:22.099.105 | C > T | p.Gln345*: sustitución de glutamina por codón de terminación prematura | Hemicigosis (1 copia) |
| 2a | |||||
| 3a | |||||
| 4b | PHEX | ChrX:22.247.942 | C > T | p.Arg747*: sustitución de arginina por codón de terminación prematura | Heterocigosis (1 copia) |
| 5b | |||||
| 6b | |||||
| 7c | PHEX | ChrX: 22.056.58 9- 22.056.590 | AG > A | p.Ser41Ilefs*5: sustitución de serina por isoleucina y cambio del marco de lectura con un consecuente codón de terminación prematura | Heterocigosis (1 copia) |
| 8c | PHEX | ChrX: 22.077.58 1- 22.077.585 | CAGAG > C | p.Arg183Serfs*37: sustitución de arginina por serina y cambio del marco de lectura con un consecuente codón de terminación prematura.Mosaicismo somático que afecta el 55% de las células somáticas | Hemicigosis (1 copia) |
Primera familia: el paciente inicial (caso1) fue confirmado genéticamente. Tenía cuatro hermanos, uno de ellos (caso2) y una hija (caso3) con el mismo diagnóstico.
Hallazgos clínicos y bioquímicos en el diagnóstico
| Caso | Edad (años) | Sexo | Signos y síntomas | Estatura | Fósforo sérico | FAL | TmP/TFG | FGF23 | |
|---|---|---|---|---|---|---|---|---|---|
| Percentil | Puntaje Z | ||||||||
| 1 | 2 | Varón | Arqueamiento de las extremidades inferiores, marcha anormal, dolor | p 1,55 | −2,16 | 3,7 | 1.712 | 1,8 | 7,78 |
| 2 | 1,5 | Mujer | Marcha anormal, ensanchamiento de las muñecas | p 22,5 | 0,75 | 2,3 | 2.346 | 1,86 | 231 |
| 3 | 1 | Mujer | Arqueamiento de las extremidades inferiores | p 50 | 0 | 3,6 | 1.760 | 3,19 | 3.106 |
| 4 | 2,75 | Mujer | Retraso en el crecimiento | p 2,31 | −2 | 3,4 | 1.295 | 2,9 | 46 |
| 5 | 2,41 | Varón | Ensanchamiento de las muñecas | p 1,86 | −2,08 | 2,6 | 422 | 2 | 66 |
| 6 | 0,75 | Varón | Retraso en el crecimiento, escafocefalia | p 0,14 | −3 | 3,5 | 907 | 3,15 | 606 |
| 7 | 1,83 | Mujer | Arqueamiento de las extremidades inferiores, marcha anormal, dolor | p 27 | −0,61 | 2,8 | 566 | 2,5 | Sin datos |
| 8 | 2 | Varón | Arqueamiento de las extremidades inferiores | p 15,6 | −1 | 2,7 | 1.897 | 2,23 | 11.358 |
FAL: fosfatasa alcalina; FGF23: factor de crecimiento fibroblástico23; TmP/TFG: reabsorción tubular máxima de fosfato por tasa de filtración glomerular.
Valores de referencia (1-3 años de edad): fósforo =3,9-6mg/dl; FAL =116-294UI/l; TmP/TFG =3,8-5mg/dl; FGF23 =0-134pg/ml.
Los casos 1 a 3 tenían la misma mutación.
Los casos 4 a 6 tenían la misma mutación.
Los casos 7 y 8 tenían mutaciones de novo.
Luego del diagnóstico, todos los pacientes recibieron terapia de reemplazo continua con 5dosis diarias de sales de fosfato (promedio: 40mg/kg/día) y 0,25μg/día de 1,25-dihidroxivitaminaD. La adherencia fue dificultosa debido a intolerancia gastrointestinal; se observó mejoría de las lesiones radiológicas de raquitismo en el primer año de tratamiento, sin normalización de los niveles de fósforo. A largo plazo, todos los pacientes presentaron signos radiológicos persistentes de raquitismo y malformaciones óseas residuales. Cuatro pacientes mantuvieron la estatura baja. Ningún paciente desarrolló hipercalciuria, nefrocalcinosis, hiperparatiroidismo ni abscesos dentales.
Debido al raquitismo persistente, el dolor osteomuscular, la limitación de las actividades diarias y las fracturas, dos niños y dos adultos fueron rotados a burosumab subcutáneo tras recibir terapia de reemplazo durante una media de 6 y 18años, respectivamente. Los niños recibieron 0,8mg/kg cada dos semanas; los adultos, 1mg/kg cada cuatro semanas, con suspensión de la terapia convencional dos semanas antes. Tras iniciar burosumab, todos los pacientes refirieron mejoría significativa en las actividades físicas, con menos fatiga y sin dolor. Se observó aumento de estatura en dos pacientes que iniciaron burosumab durante la infancia, alcanzando un puntaje Z de −0,2. Se verificó normalización del fósforo y del TmP/TFG. Los signos radiológicos de raquitismo desaparecieron en todos estos pacientes. No se observaron reacciones adversas graves.
En los pacientes con HLX, los suplementos de fosfato pueden estimular los niveles de FGF23 y la eliminación renal de fosfato, con un círculo vicioso que limita su eficacia3. La terapia convencional no es exitosa en numerosos casos, y hasta dos tercios de los niños requieren cirugías de las extremidades inferiores7. Burosumab ha demostrado ser seguro y eficaz, con mejoría de la TmP/TFG, la fosfatemia, el crecimiento lineal y la capacidad funcional, así como disminución del dolor y de la severidad del raquitismo6,8,9. Cuatro de nuestros pacientes fueron rotados a burosumab, con mejoría clínica y bioquímica y resolución completa de los signos radiológicos de raquitismo. Estos resultados fueron más notables en niños.
Concluimos que, en nuestro estudio a largo plazo y con seguimiento continuo, el hallazgo principal es la notable mejoría de las manifestaciones bioquímicas, radiológicas y especialmente clínicas en los pacientes que rotaron de tratamiento convencional a burosumab.



