


La glomerulonefritis fibrilar (GNF) es una entidad poco común definida por el hallazgo de fibrillas ordenadas al azar, no ramificadas, entre 15-25nm de diámetro en la microscopía electrónica (ME)1. Se han definido 5 patrones en la microscopía óptica (MO) relacionados con la presentación clínica y con el pronóstico2–5. Sin embargo, la mayor contribución para el diagnóstico de esta entidad ha sido el descubrimiento del marcador inmunohistoquímico DNAJB9, un marcador con una sensibilidad del 98% y una especificidad del 99%6–7. Además, el uso del marcador C4d, ampliamente utilizado en otras enfermedades renales8, puede contribuir al diagnóstico.
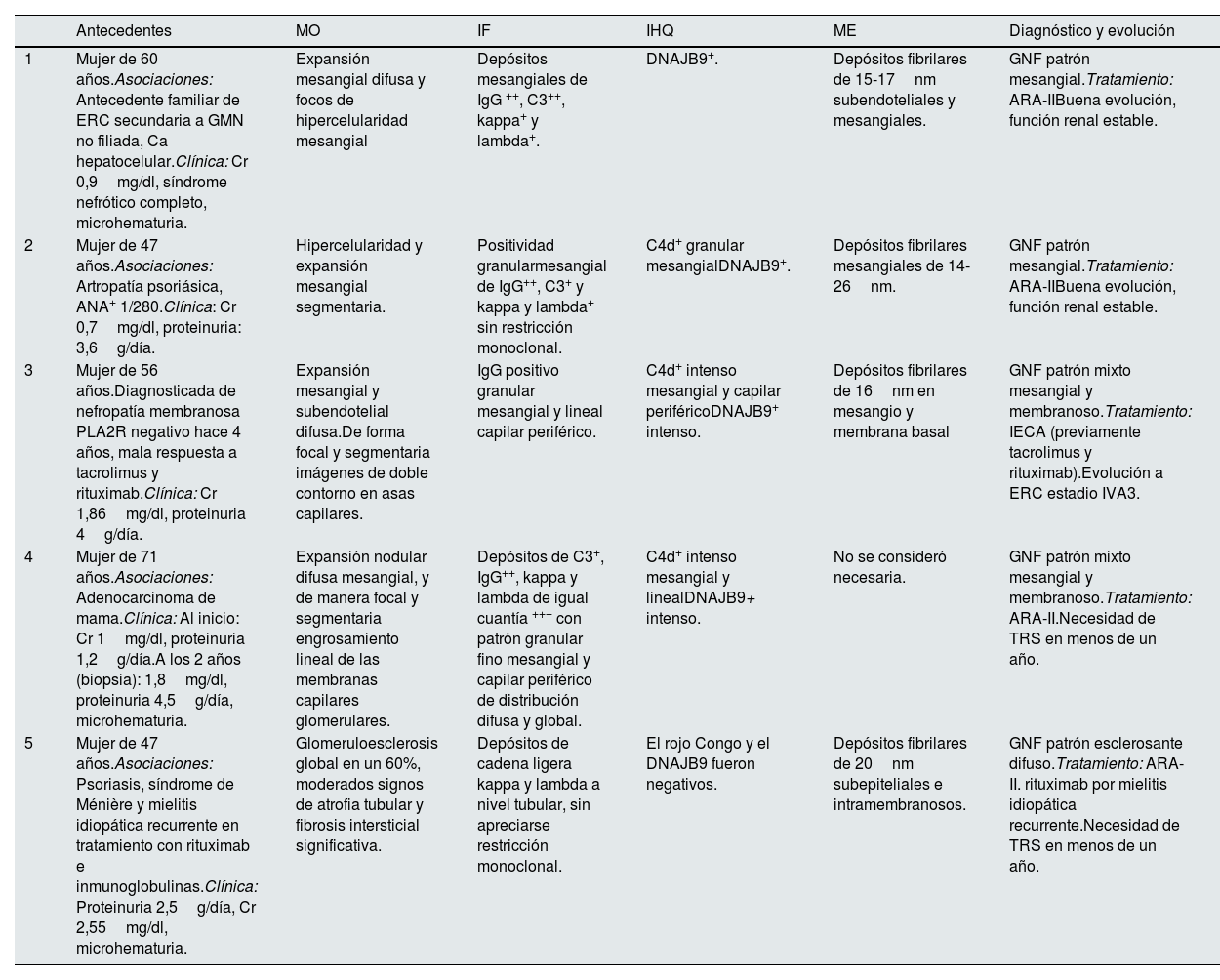
Presentamos 5 casos de GNF con relevancia epidemiológica, diagnóstica y asociativa (tabla 1 y fig. 1). La incidencia de la GNF está descrita como menor al 1%1–4, sin embargo, la incidencia de nuestra muestra es mayor, con 5 casos entre 135 biopsias realizadas, aportando una incidencia del 3,7%. Esto podría justificarse por el descubrimiento reciente del marcador DNAJB9 como herramienta diagnóstica.
Casos clínicos
| Antecedentes | MO | IF | IHQ | ME | Diagnóstico y evolución | |
|---|---|---|---|---|---|---|
| 1 | Mujer de 60 años.Asociaciones: Antecedente familiar de ERC secundaria a GMN no filiada, Ca hepatocelular.Clínica: Cr 0,9mg/dl, síndrome nefrótico completo, microhematuria. | Expansión mesangial difusa y focos de hipercelularidad mesangial | Depósitos mesangiales de IgG ++, C3++, kappa+ y lambda+. | DNAJB9+. | Depósitos fibrilares de 15-17nm subendoteliales y mesangiales. | GNF patrón mesangial.Tratamiento: ARA-IIBuena evolución, función renal estable. |
| 2 | Mujer de 47 años.Asociaciones: Artropatía psoriásica, ANA+ 1/280.Clínica: Cr 0,7mg/dl, proteinuria: 3,6g/día. | Hipercelularidad y expansión mesangial segmentaria. | Positividad granularmesangial de IgG++, C3+ y kappa y lambda+ sin restricción monoclonal. | C4d+ granular mesangialDNAJB9+. | Depósitos fibrilares mesangiales de 14-26nm. | GNF patrón mesangial.Tratamiento: ARA-IIBuena evolución, función renal estable. |
| 3 | Mujer de 56 años.Diagnosticada de nefropatía membranosa PLA2R negativo hace 4 años, mala respuesta a tacrolimus y rituximab.Clínica: Cr 1,86mg/dl, proteinuria 4g/día. | Expansión mesangial y subendotelial difusa.De forma focal y segmentaria imágenes de doble contorno en asas capilares. | IgG positivo granular mesangial y lineal capilar periférico. | C4d+ intenso mesangial y capilar periféricoDNAJB9+ intenso. | Depósitos fibrilares de 16nm en mesangio y membrana basal | GNF patrón mixto mesangial y membranoso.Tratamiento: IECA (previamente tacrolimus y rituximab).Evolución a ERC estadio IVA3. |
| 4 | Mujer de 71 años.Asociaciones: Adenocarcinoma de mama.Clínica: Al inicio: Cr 1mg/dl, proteinuria 1,2g/día.A los 2 años (biopsia): 1,8mg/dl, proteinuria 4,5g/día, microhematuria. | Expansión nodular difusa mesangial, y de manera focal y segmentaria engrosamiento lineal de las membranas capilares glomerulares. | Depósitos de C3+, IgG++, kappa y lambda de igual cuantía +++ con patrón granular fino mesangial y capilar periférico de distribución difusa y global. | C4d+ intenso mesangial y linealDNAJB9+ intenso. | No se consideró necesaria. | GNF patrón mixto mesangial y membranoso.Tratamiento: ARA-II.Necesidad de TRS en menos de un año. |
| 5 | Mujer de 47 años.Asociaciones: Psoriasis, síndrome de Ménière y mielitis idiopática recurrente en tratamiento con rituximab e inmunoglobulinas.Clínica: Proteinuria 2,5g/día, Cr 2,55mg/dl, microhematuria. | Glomeruloesclerosis global en un 60%, moderados signos de atrofia tubular y fibrosis intersticial significativa. | Depósitos de cadena ligera kappa y lambda a nivel tubular, sin apreciarse restricción monoclonal. | El rojo Congo y el DNAJB9 fueron negativos. | Depósitos fibrilares de 20nm subepiteliales e intramembranosos. | GNF patrón esclerosante difuso.Tratamiento: ARA-II. rituximab por mielitis idiopática recurrente.Necesidad de TRS en menos de un año. |
ARA-II: antagonistas de los receptores de angiotensina II; ERC: enfermedad renal crónica; GMN: glomerulonefritis; GNF: glomerulonefritis fibrilar; IECA: inhibidores de la enzima convertidora de la angiotensina; IF: inmunofluorescencia; IHQ: inmunohistoquímica; ME: microscopía electrónica; MO: microscopía óptica; TRS: tratamiento renal sustitutivo.
, DNAJB9, microscopía electrónica (ME). Caso 2: H&E, DNAJB9, C4d, ME. Caso 3: tinción metenamina de plata, C4d, DNAJB9, ME. Caso 4: H&E, tinción metenamina de plata, DNAJB9 e inmunofluorescencia IgG. Caso 5: H&E, DNAJB9 negativa, ME.")
Histología de los casos clínicos. Caso 1: hematoxilina-eosina (H&E), DNAJB9, microscopía electrónica (ME). Caso 2: H&E, DNAJB9, C4d, ME. Caso 3: tinción metenamina de plata, C4d, DNAJB9, ME. Caso 4: H&E, tinción metenamina de plata, DNAJB9 e inmunofluorescencia IgG. Caso 5: H&E, DNAJB9 negativa, ME.
Clínicamente suele comenzar a partir de la quinta-sexta década de la vida, con predominio en mujeres caucásicas, como ocurre en nuestros casos. El espectro clínico es amplio, siendo lo más frecuente la asociación de deterioro de la función renal, proteinuria y microhematuria1. En nuestra serie, 3 de las 5 pacientes presentaban función renal alterada al comienzo, todas presentaban proteinuria, siendo 4 de ellas en rango nefrótico y 3 presentaron microhematuria.
Se ha descrito su asociación con el virus de la hepatitis C, disproteinemias, enfermedades autoinmunes, diabetes mellitus y malignidad2–4. En nuestra serie, 2 pacientes presentaron enfermedad tumoral (adenocarcinoma de mama y carcinoma hepatocelular) y una paciente presentó ANA+, sin correlacionar con enfermedad autoinmune. Se describe también un caso con diagnóstico de mielitis recurrente, catalogada como idiopática, y un caso de antecedentes familiares de glomerulopatía no filiada. En la literatura, se han descrito algunos casos de asociación familiar de GNF, como el publicado en el año 2015 por Ying T et al., en el que presentaron dos familias australianas diagnosticadas GNF con patrón de herencia autosómico dominante9.
En la MO, encontramos 2 casos de patrón mesangial aislado, 2 casos con combinación de patrón mesangial y membranoso y un caso con patrón esclerosante difuso. De forma similar a la literatura1–4, los 2 pacientes con patrón mesangial presentaron mejor evolución. Dos pacientes tuvieron un patrón mixto mesangial y membranoso, que, si bien de forma aislada se han correlacionado con mejor pronóstico, la asociación de patrones podría explicar la peor evolución, especialmente para el caso número 4. Por último, la paciente con un patrón esclerosante difuso presentó mala evolución.
El reciente descubrimiento del DNJAB9 como herramienta diagnóstica6–7, con una sensibilidad del 98% y una especificidad >99%, ha supuesto un importante avance en el diagnóstico histológico, pudiendo incluso evitar la realización del estudio ultraestructural en pacientes con DNJAB9 positivo. En nuestra serie, 4 de los pacientes fueron DNAJB9 positivos, evitando el uso de la ME en uno de los casos. Para el último caso, con DNAJB9, el rojo Congo negativo descartó la posibilidad de amiloidosis evolucionada y el patrón de microscopía óptica avanzado la posibilidad de amiloidosis incipiente. Finalmente, la microscopía electrónica confirmó el diagnóstico, siendo esta la exploración gold standard.
A pesar de que el DNAJB9 es una herramienta altamente específica, no se realiza de rutina en todas las biopsias renales, por lo que es importante la sospecha histopatológica previa para solicitarlo. El uso de marcadores como el C4d, ampliamente utilizado en otras enfermedades renales, fue clave para el diagnóstico diferencial en el caso 4, previamente diagnosticada de nefropatía membranosa, ya que al observar que además del patrón clásico de depósito membranoso, también había depósito mesangial, lo que hizo plantearse otros diagnósticos, solicitar el DNAJB9 y la ME, y confirmar así el diagnóstico.
No se ha establecido una terapia óptima para la glomerulonefritis fibrilar hasta la fecha1. Además de la terapia médica conservadora para el tratamiento de la proteinuria, se han ensayado con algunos inmunosupresores: esteroides, micofenolato, rituximab o ciclofosfamida. Sin embargo, los resultados no son muy alentadores, siendo el rituximab el que ha mostrado un aparente beneficio10. En nuestra serie, todas las pacientes se trataron con manejo conservador tras la confirmación diagnóstica. Además, 2 pacientes recibieron tratamiento con rituximab, una por el diagnóstico previo de nefropatía membranosa y otro caso como tratamiento de su mielopatía recurrente. Ambas presentaron mala evolución.
En conclusión, la GNF es una entidad clínica poco frecuente, pero probablemente está infradiagnosticada. La aparición de nuevas herramientas diagnósticas, como el marcador DNAJB9, puede permitir que en los próximos años el número de diagnósticos se incrementen. El pronóstico se relaciona con el patrón histológico observado, y en la actualidad, las opciones terapéuticas son limitadas. Por tanto, se requiere llevar a cabo nuevos estudios para mejorar el tratamiento y la gestión de la enfermedad.



