








La glomerulonefritis rápidamente progresiva constituye un síndrome clínico que se manifiesta con signos de daño glomerular a nivel urinario junto con un deterioro progresivo de la función renal más o menos rápido. Se clasifica en cinco tipos atendiendo a su base etiopatogénica. Se presentan dos casos de glomerulonefritis rápidamente progresiva llamativos por su forma de presentación y evolución. Se detallan los aspectos que motivaron nuestro manejo diagnóstico y terapéutico, así como algunos relativos al tratamiento integral de esta entidad nosológica.
INTRODUCCIÓN
Según el registro de glomerulonefritis de la Sociedad Española de Nefrología (S.E.N.), la disfunción renal aguda es la tercera causa de biopsia renal hasta el año 2012. Histológicamente, el lupus eritematoso sistémico (LES) representa la principal entidad asociada a afectación multisistémica en la edad adulta (15-65 años).
A continuación se exponen dos casos de glomerulonefritis rápidamente progresiva con afectación multisistémica, que debutaron con cuadros clínicos similares, pero con etiopatogenia y evolución muy diferentes.
CASOS CLÍNICOS
Caso 1
Paciente de 20 años, hispana, sin antecedentes de interés, atendida en urgencias tras cuadro diarreico de 3 días de evolución (tres deposiciones/día) y epigastralgia. A la exploración física, la paciente presentaba: presión arterial de 153/92 mmHg; frecuencia cardíaca de 112 lat/min; temperatura de 37,9 ºC; SO2 del 95 %; sin signos de deshidratación ni otras alteraciones. Analíticamente se identificó anemia normocítica y elevación de la creatinina sérica habitual (de 0,6 a 1,9 mg/dl). Las pruebas de imagen demostraban un derrame pleural leve bilateral, aumento de la ecogenicidad renal bilateral y líquido libre peritoneal en escasa cuantía, sin otros hallazgos.
A las 24 h del ingreso, la hemoglobina era de 7,9 g/dl, sin evidencia de hemorragia y asociada a un descenso plaquetario. La extensión sanguínea objetivó un 3 % de esquistocitos, con Coombs directo positivo. Las cifras de creatinina aumentaron hasta 2,5 g/dl. Otros datos relevantes del estudio fueron la presencia de títulos bajos de C3 y C4, actividad ADAMTS13 (A Disintegrin-like And Metalloprotease with ThromboSpondin type 1 motif, member 13) normal, anticuerpos antinucleares, anti-ADN, antihistonas y antinucleosomas positivos, con anticuerpos anti-fosfolípido negativos (tabla 1). Los resultados de hemocultivos, urocultivos (incluidos parásitos) y coprocultivos (incluidos parásitos y cepas de Escherichia coli enteroinvasivas) fueron negativos.
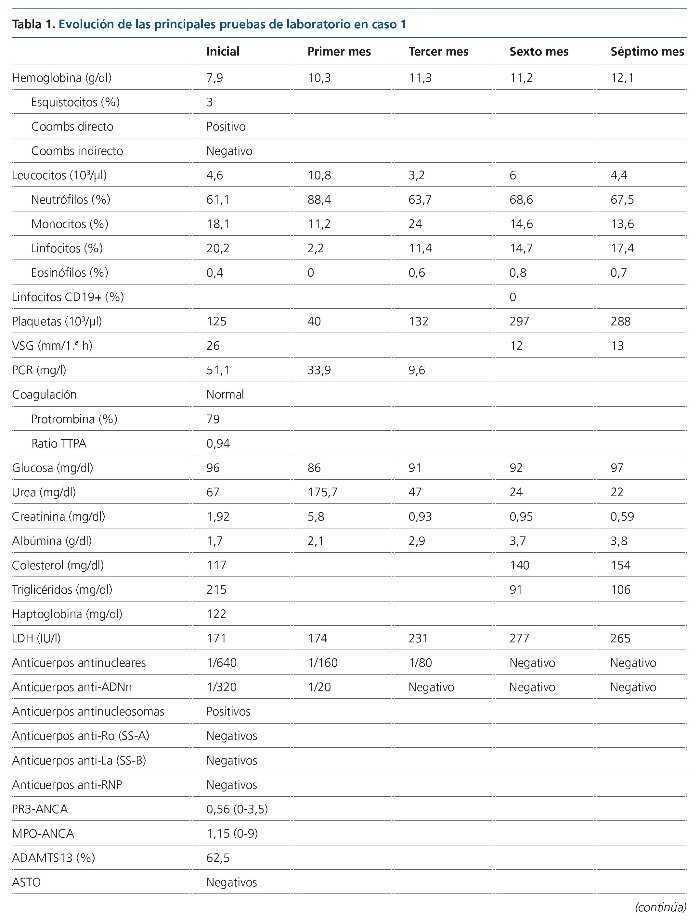
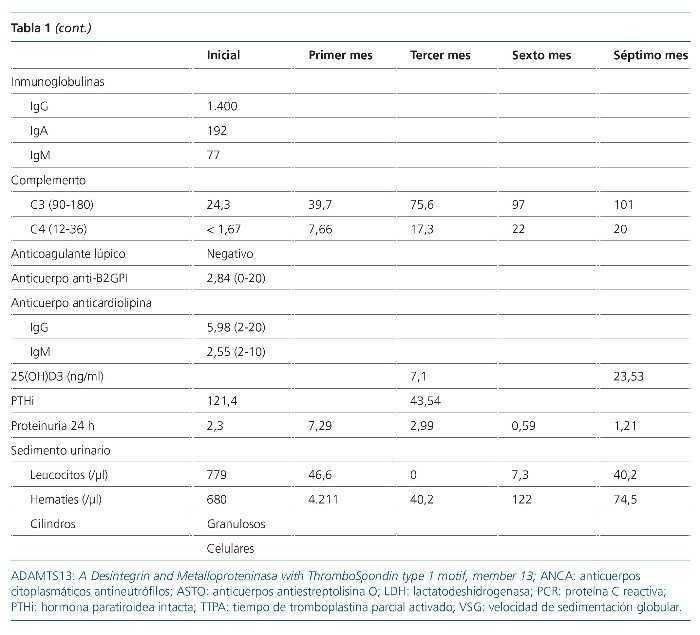
A las 72 h del ingreso se llevó a cabo una biopsia renal y se instauró tratamiento con pulsos intravenosos de 1.000 mg de metilprednisolona, continuado de prednisona a 0,5 mg/kg/día. El estudio anatomopatológico (55 glomérulos) mostró signos de nefropatía lúpica tipo IV-G (A), con un índice de actividad 15/24 y un índice de cronicidad 0/12 (figura 1). Teniendo presentes la gravedad de la nefropatía y la raza de la paciente se decidió instaurar tratamiento de primera línea con ciclofosfamida intravenosa a dosis altas (750 mg/m2).
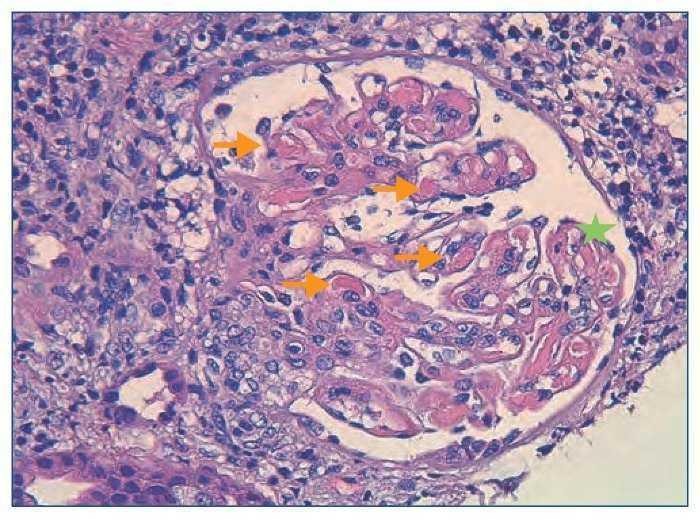
Figura 1. Histología renal caso 1 (tinción hematoxilinaeosina).
Glomérulo con engrosamiento de las paredes capilares (estrella) y presencia de trombos hialinos intracapilares característicos de la microangiopatía trombótica (flechas).
La evolución en las semanas posteriores fue desfavorable, con progresión de la disfunción renal hacia oliguria e hiperpotasemia, por lo que se inició tratamiento hemodialítico. Desde el punto de vista hematológico mantuvo una tendencia a la anemización y la trombopenia; precisó la transfusión de hasta seis concentrados de hematíes y alcanzó títulos de 40.000/µl plaquetas. El complemento tampoco aumentó. Todo ello habiendo aumentado la dosis de esteroides a 1 mg/kg/24 h. Sin más dilación, transcurridas 4 semanas de la administración de la primera dosis de ciclofosfamida, se hizo una conversión del tratamiento inmunosupresor a micofenolato (MMF: 2,5 g/24 h) asociado a rituximab (375 mg/m2/semana, cuatro dosis).
Posteriormente, la evolución mejoró notablemente, con recuperación de la función renal hasta aclaramiento de creatinina de 158,83 ml/min, disminución de la proteinuria de 7,29 a 1,21 g/24 h y de la hematuria de > 2.000/µl a 74,5 hematíes/µl. Desde el punto de vista hematológico se corrigieron la trombopenia y la anemia. Los títulos de ANA y Anti-ADNn se negativizaron y los valores de complemento se normalizaron. Por tanto, se obtuvo una remisión parcial y la paciente mantuvo unos valores de CD19+ indetectables a los 6 meses del tratamiento con rituximab.
Añadir que desde el inicio del tratamiento se tomaron medidas profilácticas frente a posibles complicaciones derivadas de las terapias instauradas (cotrimoxazol, carbonato de calcio, vitamina D y las vacunaciones pertinentes según programa de inmunización recibido previamente por la paciente y necesidades atribuibles al tratamiento inmunosupresor). Un control densitométrico (DEXA) a los 4 meses objetivaba signos de osteopenia, por lo que se inició tratamiento con teriparatida.
Caso 2
Paciente de 34 años de edad, hispana, con antecedentes personales de anemia ferropénica, bocio nodular adenomatoide con hipotiroidismo en tratamiento sustitutivo, que ingresa en el servicio de medicina interna por cuadro de tendinitis aquilea bilateral de 15 días de evolución, sucedida de un cuadro de faringoamigdalitis, con fiebre y adenopatías cervicales. Se instauró tratamiento con amoxicilina-clavulánico, antiinflamatorios, hierro y vitamina B1-B6-B12. Posteriormente aparecieron signos de artritis en antepiés, anemización (Coombs directo positivo), hematuria y elevación de las cifras de creatinina sérica (de 0,89 a 2,5 mg/dl). Con tratamiento esteroideo a dosis bajas (prednisona 10 mg/24 h) desaparecieron los signos artríticos, pero no el dolor en extremidades inferiores. Un electromiograma posterior demostró la presencia de una neuropatía axonal sensitiva de extremidades inferiores.
En este contexto se amplió el estudio para descartar la presencia de una patología autoinmune sistémica, y se identificó normocomplementemia, títulos de MPO-ANCA elevados (tabla 2) y biopsia renal (ocho glomérulos) compatible con glomerulonefritis rápidamente progresiva pauciinmune (figuras 2 y 3). Así pues, el estudio se concluyó con el diagnóstico de poliangeítis microscópica (PAM) generalizada (BVAS 3.0: 21; VDI: 3) y se inició un tratamiento de inducción de la remisión con ciclofosfamida intravenosa (15 mg/kg administrados cada 4 semanas, durante 6 meses), junto a tres dosis de 1.000 mg de metilprednisolona intravenosa, sucedidas de prednisona oral (1 mg/kg/día).
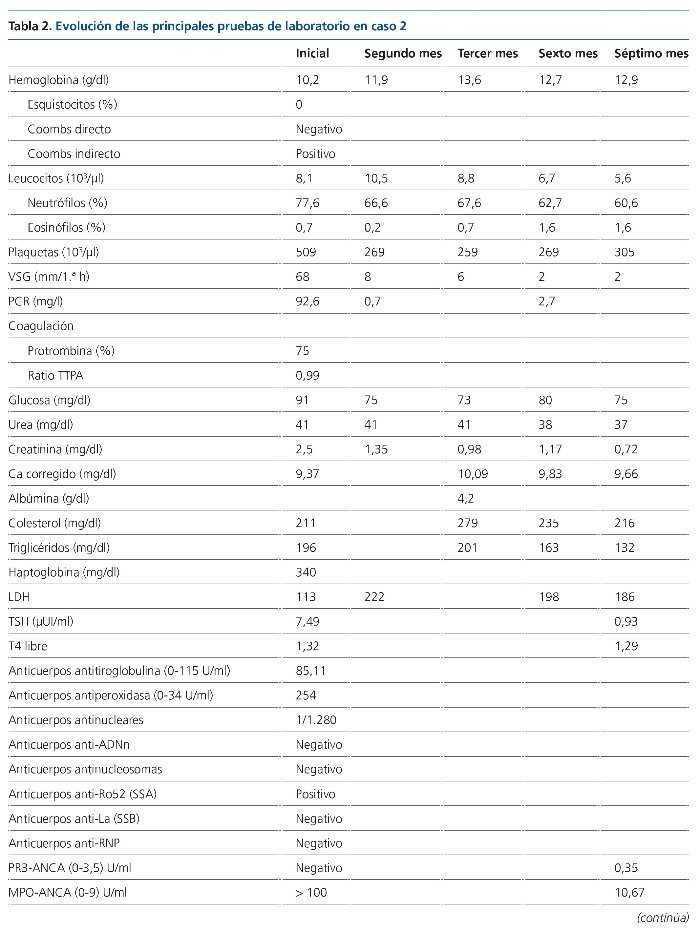
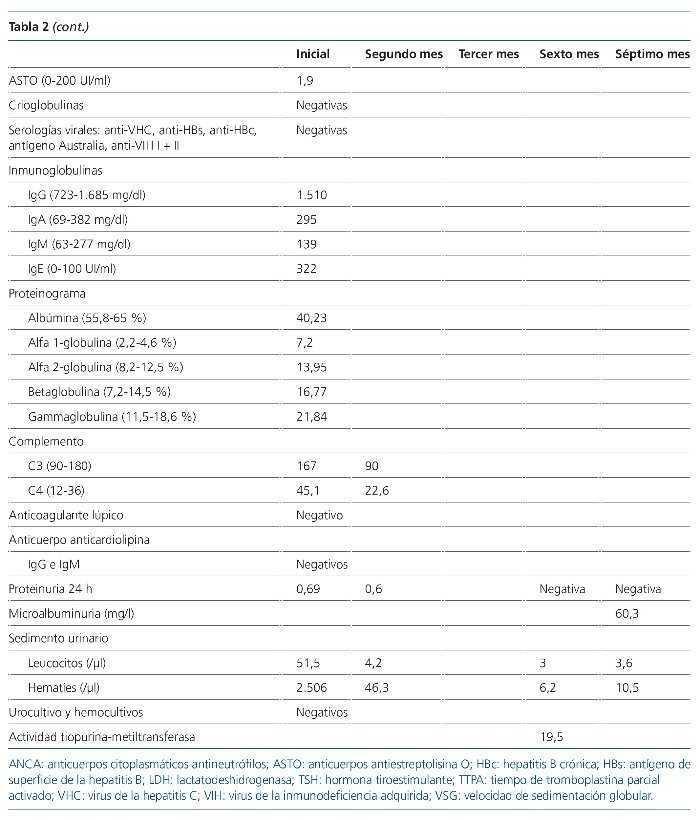
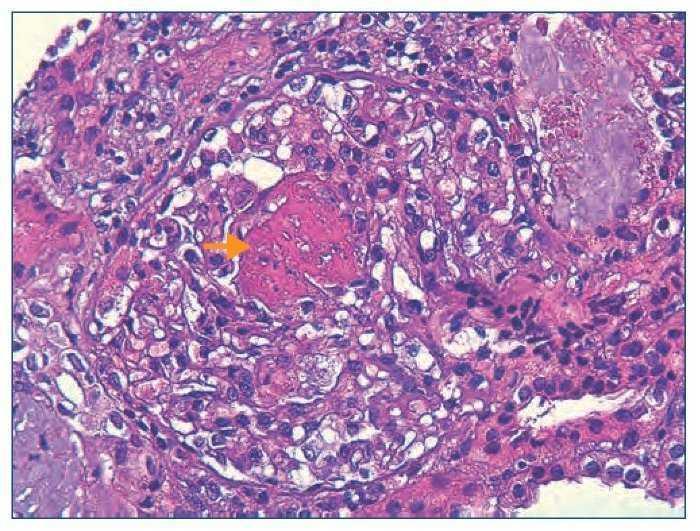
Figura 2. Histología renal caso 2 (tinción hematoxilinaeosina).
Necrosis fibrinoide del penacho glomerular (flecha) característica de la glomerulitis asociada a las vasculitis de pequeño vaso.
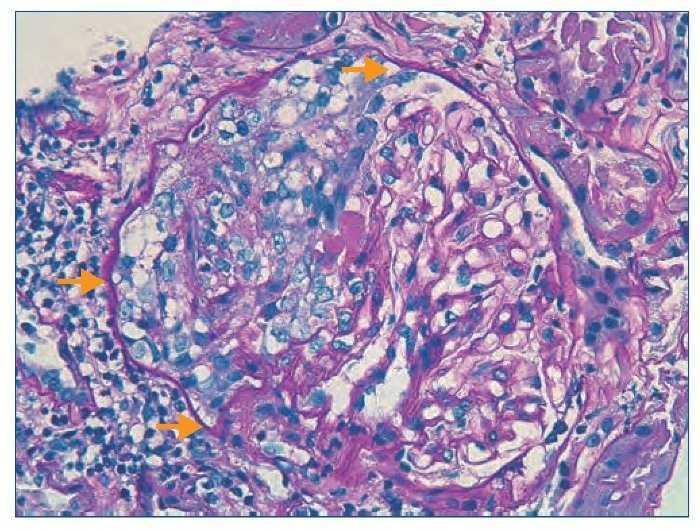
Figura 3. Histología renal caso 2 (tinción con ácido periódico de Schiff).
Semiluna celular (flechas) característica de las glomerulonefritis rápidamente progresivas en una fase activa y aguda.
La evolución posterior fue favorable, con una recuperación rápida de la función renal, normalización del sedimento urinario, corrección de la anemia y desaparición lentamente progresiva de los síntomas secundarios a la neuropatía periférica. En el momento actual, la paciente se encuentra en remisión completa y se ha iniciado tratamiento de mantenimiento con azatioprina a dosis de 2 mg/kg/día, junto a dosis descendentes de prednisona. Igualmente se tomaron medidas profilácticas (mesna, cotrimoxazol, carbonato de calcio y vitamina D, leuprorelina, calendario de vacunaciones). Ante la presencia de osteopenia en DEXA, también se inició teriparatida.
DISCUSIÓN
La glomerulonefritis rápidamente progresiva es un síndrome clínico que se manifiesta por signos de daño glomerular a nivel urinario, junto con un deterioro progresivo de la función renal, en un período que puede comprender desde días hasta semanas o meses.
Histológicamente se caracteriza por el desarrollo de semilunas epiteliales glomerulares. Fisiopatológicamente, el daño glomerular se origina en las paredes de los capilares glomerulares, lo cual induce una respuesta inflamatoria a nivel del espacio de Bowman y finalmente conducirá al desarrollo de semilunas fibroepiteliales y fibrosas. Estas últimas constituyen un estadio avanzado de lesión glomerular, con pocas posibilidades de regresión1.
Atendiendo a los mecanismos etiopatogénicos se clasifican como:
• Tipo 1: por presencia de anticuerpos antimembrana basal glomerular (enfermedad de Goodpasture).
• Tipo 2: por depósito de inmunocomplejos circulantes en el seno de patologías como la nefropatía IgA, nefropatía membranosa, glomerulonefritis postinfecciosa, nefropatía lúpica o crioglobulinemia mixta esencial.
• Tipo 3 o pauciinmune: no hay presencia de depósitos inmunes bajo el estudio de inmunofluorescencia o de microscopia electrónica. La mayoría de los pacientes presentan títulos de ANCA elevados.
• Tipo 4: por presencia de anticuerpos doblemente positivos (características asociadas de los tipos 1 y 3).
• Idiopática: es un término aplicado en dos contextos, el tipo 2 que no encaja con ninguna de las categorías identificadas y el tipo 3, ANCA-negativo.
Clínicamente puede ponerse de manifiesto de forma multisistémica o existir una afectación limitada al riñón. Los dos casos presentados corresponden a dos mujeres jóvenes, que debutaron con un cuadro de disfunción renal subaguda asociada a manifestaciones sistémicas.
El caso 1, con un cuadro grave de anemia hemolítica, trombopenia y afectación renal precedido por un episodio de diarreas, por lo que se realizó un estudio basado en las recomendaciones para el diagnóstico de la microangiopatía trombótica del documento de consenso de la S.E.N.2. Los estudios complementarios revelaron, en primer lugar, una actividad ADAMTS13 normal, lo cual descartaba la presencia de una púrpura trombótica trombocitopénica, con coprocultivo para bacterias enteropatógenas negativo (cribado de síndrome hemolítico urémico típico) y hallazgo analítico de títulos elevados de anticuerpos anti-ADNn, entre otros. El resultado histológico de la biopsia renal, finalmente, confirmó el diagnóstico de nefropatía lúpica tipo IV-G (A) y la paciente fue diagnosticada de LES, con microangiopatía trombótica secundaria. En cuanto al tratamiento, se inició con ciclofosfamida, pues existe una recomendación más fuerte que para MMF en los pacientes con afectación renal grave. Entre los esquemas de tratamiento con ciclofosfamida, aunque más seguro, descartamos el de dosis bajas (Euro-Lupus Nephritis Trial), por no estar demostrada su eficacia en pacientes con afectación renal grave o latinoamericanos3,4.
El caso 2 resulta llamativo, ya que las vasculitis ANCA-positivo tienen su pico de incidencia situado entre los 65-74 años y es más frecuente en la raza caucásica (98 % de los casos)5. Nuestra orientación diagnóstica inicial, habiendo descartado signos de endocarditis, fue de LES. Sin embargo, los títulos normales de complemento no eran compatibles con este y, posteriormente, la positividad de los anticuerpos MPO-ANCA, junto con los hallazgos histológicos, fue determinante para el diagnóstico de un proceso vasculítico de pequeño vaso. Sobre la base del algoritmo diagnóstico de las vasculitis ANCA-positivo y la PAN de Watts et al6 le diagnosticamos PAM (afectación renal y extrarrenal, hallazgos histológicos de vasculitis de pequeño vaso, títulos elevados de p-ANCA y sin clínica sugestiva de enfermedad de Churg-Strauss o marcadores subrogados de granulomatosis con poliangeítis). La paciente cumplía los criterios de la European League Against Rheumatism7 de afectación generalizada con scores de actividad y daño derivado de la enfermedad significativos8, por lo que optamos por una estrategia inmunosupresora potente (ciclofosfamida), la cual se administró de forma intravenosa por estar asociada a una tasa inferior de efectos secundarios (estudio CYCLOPS)9.
La evolución tras el tratamiento fue diferente en ambos casos. El caso 1 evolucionó de forma desfavorable, continuando el proceso de anemización, la trombopenia y la progresión del daño renal. Por este motivo, bajo las recomendaciones de las guías3,4, se optó por conversión a inmunosupresión con MMF, pero dada la gravedad de la nefropatía se decidió intensificarla con rituximab. Afortunadamente, en pocas semanas, la paciente obtuvo una remisión parcial, que ha ido mejorando progresivamente, aunque sin ser completa. En el caso 2, sin embargo, la paciente, también de raza hispana, obtuvo una remisión completa con ciclofosfamida intravenosa (BVAS 3.0: 6; VDI: 1, tras 6 meses).
Tanto el LES como la PAM son enfermedades sistémicas de base autoinmune. Disponemos de un arsenal terapéutico similar para el abordaje de ambas entidades, fundamentalmente ciclofosfamida, MMF, azatioprina y rituximab. Sin embargo, responden de forma diferente frente a este. Así, en el LES, MMF es un tratamiento de primera línea, tanto en la inducción como en el mantenimiento, aunque todavía no está aclarada su eficacia en casos con creatinemia > 3 mg/dl o hallazgos histológicos de necrosis fibrinoide o semilunas. Por el contrario, en la PAM son necesarios más estudios que justifiquen su empleo como terapia de inducción, y en terapia de mantenimiento sería un fármaco de segunda o tercera línea, por detrás de azatioprina (primera línea de mantenimiento) y posiblemente de metrotexato10-12.
Rituximab, en el LES no ha demostrado utilidad en la inducción (ensayos EXPLORER y LUNAR). Su indicación actual es para pacientes con múltiples recaídas o no respondedores (nivel de evidencia 2B), y su combinación con MMF es beneficiosa, sin exceso de toxicidad. Nuestra experiencia en el caso 1 nos motiva a replantear su utilidad combinado con MMF en pacientes con afección renal grave, incluidos los de raza afroamericana. En cambio, en las vasculitis se considera un fármaco de primera línea en inducción, sin inferioridad a la ciclofosfamida (ensayos RAVE y RITUXVAS)12,13.
Por otra parte, queremos referirnos al estudio de la tiopurinmetiltransferasa (TPMT) previa al tratamiento con tiopurinas (azatioprina), que llevamos a cabo en el caso 2. La TPMT es una enzima encargada de metabolizar las tiopurinas, dando lugar a metabolitos derivados de la tioguanina, cuyo acúmulo se relaciona tanto con el riesgo de mielotoxicidad como con el control de la enfermedad. La actividad de la TPMT está regulada por polimorfismos genéticos, y se ha identificado un 11 % de individuos heterocigotos para un alelo de baja actividad (implica una actividad intermedia y en 1 de cada 300 individuos una ausencia total de actividad). El British National Formulary y la Federal Drug Administration recomiendan el estudio de su fenotipo y/o genotipo previo al inicio de tratamientos con tiopurinas, y hay estudios que demuestran que es una medida coste-eficiente14,15. Hasta la fecha, el estudio de TPMT ha sido implementado sobre todo en dermatología, reumatología y gastroenterología. En las vasculitis, tan solo un estudio retrospectivo evaluó la utilidad de la determinación del genotipo y fenotipo, sin concluir en su eficacia16. No obstante, dados los resultados obtenidos en otras entidades nosológicas, consideramos que son necesarios estudios que aporten mayor nivel de evidencia sobre esta cuestión. En cualquier caso, el estudio de TPMT no excluye la necesidad de llevar a cabo una monitorización del hemograma.
Finalmente, hacer referencia a la idoneidad de instaurar otras medidas profilácticas frente a los posibles efectos secundarios derivados del uso de inmunosupresores. Con ciclofosfamida: cotrimoxazol para la profilaxis de la infección por Pneumocystis jiroveci; MESNA para disminuir el riesgo de toxicidad vesical; leuprorelina para prevenir la amenorrea en mujeres con edad superior a 35 años o previsión de dosis acumuladas > 10 g. En pacientes que van a recibir tratamiento con dosis de prednisona > 5-7,5 mg/24 h por más de 3 meses, medidas de protección ósea: carbonato de calcio y vitamina D, junto a una monitorización densitométrica ósea; en pacientes de riesgo o con desarrollo de osteopenia-osteoporosis está indicado el uso de bifosfonatos (alendronato, risendronato o ácido zoledrónico) o teriparatida17. Otras medidas son la protección gástrica (pacientes con uso concomitante de esteroides y AINE [antiinflamatorios no esteroideos]) y el control de los factores de riesgo cardiovascular.
CONCLUSIONES
La glomerulonefritis rápidamente progresiva puede tener múltiples etiologías. Clínicamente puede existir un solapamiento importante entre las distintas etiologías, por lo que es fundamental completar su estudio con datos de laboratorio e histológicos. Pese a disponer de similares estrategias terapéuticas, existen diferencias en cada una de estas patologías a tener en consideración. Además, todavía existen cuestiones al respecto pendientes de resolver. Así, en pacientes con LES y nefropatía grave, de raza hispana, ¿podría tener un papel en la inducción el tratamiento combinado de rituximab y MMF frente a ciclofosfamida?
Otro aspecto importante, en cuanto al manejo de estos tratamientos, es la prevención de complicaciones derivadas de estos. En este sentido, la utilidad de estrategias como la determinación de la actividad de TPMT aunque prometedora, debe ser mejor estudiada.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Correspondencia: Sara Bielsa-Gracia
Servicio de Nefrología.
Hospital Obispo Polanco.
Avda. Ruiz Jarabo, s/n. 44002 Teruel.
bielsagracia@hotmail.com


