



La glomeruloesclerosis focal y segmentaria (GEFS) es un patrón histológico de lesión que deriva de diversos procesos patológicos que afectan a los podocitos, resultando en pérdida de selectividad del filtrado glomerular, proteinuria y desarrollando insuficiencia renal que progresa a enfermedad renal crónica terminal en un importante número de pacientes. La clasificación propuesta por las guías KDIGO, en 2021, divide la GEFS en cuatro categorías: primaria, secundaria, genética y de causa no determinada, facilitando así su diagnóstico y manejo. Las causas hereditarias de la GEFS presentan una variabilidad clínica significativa, complicando su identificación. El estudio genético es crucial para identificar la GEFS de causa genética. La prevalencia de la GEFS genética es significativa en niños y considerable en adultos, destacando la importancia del diagnóstico temprano para evitar tratamientos innecesarios y facilitar el consejo genético. Las técnicas de secuenciación masiva han revolucionado el diagnóstico genético, permitiendo la identificación de más de 60 genes responsables del daño podocitario. Este documento propone recomendaciones clínicas y patológicas para la realización de estudios genéticos en adultos con GEFS, subrayando la necesidad de una correcta clasificación para la planificación terapéutica adecuada y la mejora de los resultados en ensayos clínicos.
Focal segmental glomerulosclerosis (FSGS) is a histological pattern of injury that derives from various pathological processes that affect podocytes, resulting in loss of selectivity of the glomerular filtration membrane, proteinuria and the development of renal failure that progresses to end-stage kidney disease in a significant number of patients. The classification proposed by the 2021 KDIGO guidelines divides FSGS into four categories: primary, secondary, genetic, and FSGS of undetermined cause, thus facilitating its diagnosis and management. Genetic causes of FSGS present significant clinical variability, complicating their identification. Genetic testing is crucial to identify FSGS of genetic cause. The prevalence of genetic FSGS is significant in children and considerable in adults, highlighting the importance of early diagnosis to avoid unnecessary treatments and facilitate genetic counselling. Massive sequencing techniques have revolutionized genetic diagnosis, allowing the identification of more than 60 genes responsible for podocyte damage. This document proposes clinical recommendations for carrying out genetic studies in adults with FSGS, highlighting the need for a correct classification for adequate therapeutic planning and improvement of results in clinical trials.
Se debe entender la glomeruloesclerosis focal y segmentaria (GEFS) como un patrón histológico, no como una enfermedad. Este patrón histológico es el resultado de una variedad de procesos patológicos que comparten un daño común sobre los podocitos; supone la pérdida de la selectividad del filtrado glomerular y clínicamente se traduce en la aparición de proteinuria e insuficiencia renal a largo plazo1. Histológicamente, la pérdida podocitaria supone la migración de células epiteliales de la cápsula de Bowman para intentar sustituir a los podocitos; sin embargo, este intento de regeneración es ineficiente y produce esclerosis mesangial parcheada del penacho glomerular (segmentaria) que comienza en algunos glomérulos pero no en todos (focal)2. Las guías KDIGO (Kidney Disease: Improving Global Outcomes) para el manejo de las enfermedades glomerulares, publicadas en 2021, proponen una nueva clasificación, etiopatogénica y dividida en cuatro categorías, lo que facilita el diagnóstico y abordaje terapéutico. Así, tenemos formas primarias, secundarias, genéticas o de causa no determinada3. Los antecedentes familiares, personales y la forma de presentación clínica no siempre son suficientes para excluir una causa hereditaria. La presentación clínica de las GEFS de causa genética es extremadamente variable; con diferencias en la edad de debut, grado de proteinuria y la forma de progresión de la enfermedad renal crónica (ERC). Inicialmente, las formas genéticas fueron descritas con un comienzo en la edad infantil, asociadas principalmente a síndrome nefrótico resistente a corticoides; sin embargo, en función de los criterios de selección, hasta un 30% de los casos de GEFS en adultos puede estar asociado a una causa genética4–8, por lo que sigue siendo un reto definir los criterios para realizar estudio genético. Las características clínicas e histológicas que parecen predecir mejor la etiología genética son: la ausencia de respuesta a las terapias inmunosupresoras, la presencia de microhematuria, la ausencia de fusión pedicelar difusa en la biopsia renal y mantener una albúmina sérica normal a pesar de desarrollar una proteinuria en rango nefrótico6,9,10, aunque estas últimas dos características pueden modificarse según el momento del diagnóstico.
Está bien establecido que reconocer el diagnóstico de la GEFS de causa genética es de vital importancia para los pacientes. Por una parte, el diagnóstico precoz evita exploraciones y exposición a tratamiento inmunosupresor innecesario y permite, además, el diagnóstico de portadores asintomáticos; despistaje de patologías asociadas y facilita el consejo genético. A diferencia de las formas primarias de la GEFS, su recidiva de causa genética en el trasplante renal no es un problema; pero sí es relevante en el desarrollo de otras patologías, como anticuerpos anti-membrana basal glomerular, que ocurre en el 2-3% de los pacientes portadores de un trasplante renal con síndrome de Alport ligado al cromosoma X11. Finalmente, también permite realizar una selección adecuada de potenciales donantes renales vivos emparentados.
Una correcta clasificación de los pacientes según sus características clínicas e histológicas es fundamental para la toma de decisiones y la planificación de un adecuado esquema terapéutico. Desafortunadamente, muchos ensayos clínicos han fracasado por incluir pacientes con distintas formas de GEFS sin una correcta estratificación.
En el presente documento proponemos unas recomendaciones basadas en criterios clínicos y anatomopatológicos para la realización de estudio genético en pacientes adultos con GEFS.
Glomeruloesclerosis focal y segmentaria primariaLa etiología de la GEFS de causa inmunológica, clásicamente llamada «GEFS primaria», aún no está del todo aclarada. En las últimas décadas se ha supuesto que es causada por un factor de permeabilidad circulante, que se especula que consiste en un grupo de citoquinas que alteran de forma brusca la función podocitaria, aumentando la permeabilidad de la membrana de filtración glomerular. Este grupo de citoquinas aún no ha sido definido, y existen varias moléculas que se han propuesto como posibles causantes de la enfermedad, entre las que se incluyen el factor 1 de citoquina similar a cardiotrofina (CLCF-1), el receptor soluble del activador de plasminógeno tipo uroquinasa (suPAR), el anticuerpo anti-CD40, la apolipoproteina A1 y la forma soluble de proteína quinasa calcio/calmodulina-serina (CASK)12. En los últimos años se ha identificado que una proporción de pacientes con enfermedad de cambios mínimos son causados por los anticuerpos antinefrina13, y en trabajos posteriores se ha confirmado su papel en la GEFS primaria, especialmente en las formas recidivantes tras el trasplante14. Sin embargo, recientemente se ha identificado que solamente un 9% de los pacientes con GEFS primarias son causadas por estos anticuerpos15, por lo que queda por esclarecer cuáles son los agentes implicados en ese factor de permeabilidad que contribuye al resto de casos de GEFS.
La GEFS primaria se presenta de forma brusca y en forma de síndrome nefrótico completo (proteinuria ≥3,5g/día, hipoalbuminemia, hipercolesterolemia y edema). Histológicamente en el microscopio óptico no existen características específicas que la distingan de las otras formas de GEFS, mientras que en el microscopio electrónico destaca la presencia de una fusión pedicelar difusa que ocupa más del 80% de la superficie de la membrana glomerular y la diferencia de las formas secundarias, siendo un hallazgo determinante. En una serie de pacientes adultos y predominantemente blancos con GEFS primaria, la mediana de fusión pedicelar fue del 100%16; sin embargo, hay que tener en cuenta que varias formas hereditarias de GEFS pueden presentar también una fusión pedicelar difusa. Un análisis morfométrico del ancho de los pedicelos, que excluyó a los pacientes con formas hereditarias de GEFS, encontró pedicelos más amplios en pacientes con GEFS primaria en comparación con aquellos con GEFS secundaria. Una anchura pedicelar >1.500nm diferenciaba adecuadamente la GEFS primaria de la secundaria17. Hallazgos histológicos, como la vacuolización de los podocitos y la transformación microvellositaria, están relacionados con la cuantía de la proteinuria y, aunque inespecífico, es más frecuente visualizarlo en las formas primarias que en las secundarias18.
El tratamiento de la GEFS primaria es la inmunosupresión, y debido a su mal pronóstico en ausencia de tratamiento se recomienda iniciar precozmente la terapia inmunosupresora en casos de GEFS que debutan como síndrome nefrótico completo19.
El tratamiento con glucocorticoides es la piedra angular actual del manejo de la GEFS primaria3; Sin embargo, esta recomendación se basa en estudios observacionales y no existen estudios que hayan comparado la prednisona con placebo en el tratamiento de la GEFS primaria20. Un pequeño ensayo clínico observó que la asociación de micofenolato a dosis más bajas de esteroides conseguía resultados similares con una dosis acumulada de esteroides menor21.
La respuesta a la terapia con corticoides es una característica definitoria de la GEFS primaria, que descartaría otras causas de GEFS, aunque en ocasiones la definición de la resistencia a glucocorticoides es difícil. Estudios recientes han llegado a demostrar una variante genética en hasta un 42% de los pacientes con resistencia a glucocorticoides22, por lo que la realización de un estudio genético estaría especialmente justificada en este grupo de pacientes para evitar el tratamiento innecesario con inmunosupresores.
Los inhibidores de la calcineurina (ICN) son el tratamiento estándar para pacientes con contraindicaciones, intolerancia o resistencia a los glucocorticoides3. El mecanismo de acción de los glucocorticoides y los ICN en la GEFS primaria no está del todo claro; se presupone que estas terapias interfieren con los sitios celulares en los que se producen los supuestos factores de permeabilidad. Sin embargo, la información es un tanto ambigua sobre si el beneficio de los inmunosupresores depende únicamente de su efecto inmunosupresor o si se debe a un efecto local sobre la función de los podocitos y la estabilización de la sinaptopodina23. Las diferentes series han descrito una tasa de respuesta a glucocorticoides y/o ICN en la GEFS en el 40-70% de los casos19, aunque esta proporción probablemente esté infraestimada debido a la inclusión de pacientes con formas secundarias o hereditarias dentro de estos estudios y probablemente la tasa de respuesta a terapia inmunosupresora adecuado sea mayor en pacientes correctamente clasificados como GEFS primaria.
Una vez descartadas las causas genéticas y secundarias, la tasa de recurrencia de la GEFS primaria en el trasplante renal es de hasta un 80% en primeros trasplantes24. La recurrencia precoz en el trasplante es el sello distintivo de la GEFS primaria. No se conoce aún cual es el mejor tratamiento para la GEFS recurrente en el trasplante. La plasmaféresis profiláctica no reduce el riesgo de recurrencia, aunque la plasmaféresis terapéutica se considera como una buena opción de primera línea como terapia para eliminar los factores de permeabilidad, especialmente en recurrencias muy tempranas con proteinuria severa25. El tratamiento profiláctico con rituximab no ha demostrado ser eficaz para reducir el riesgo de recurrencia26.
Glomeruloesclerosis segmentaria y focal secundariaLa mayoría de las formas secundarias de GEFS son debidas a una mala adaptación entre la carga y la capacidad glomerular. El mecanismo patogénico es siempre común y consiste con el daño y pérdida podocitaria adaptativa secundaria al incremento de la demanda sobre los mismos (incapacidad para adaptarse al tamaño glomerular, isquemia debida a hipoperfusión). Esto puede ocurrir por circunstancias que reducen la masa renal, como la baja masa nefronal al nacer, la nefropatía por reflujo, la displasia renal o situaciones en la que existe un aumento en la tasa de filtración glomerular que sobrepasa la capacidad glomerular, como en la obesidad, la ingesta elevada de proteínas o el abuso de andrógenos27–29. Cualquier enfermedad glomerular o tubular crónica puede reducir la función total de la nefrona y provocar una GEFS maladaptativa que se superpone al trastorno primario30.
La hiperfiltración y la hipertensión en el capilar glomerular representan el mayor mecanismo de tensión sobre el podocito. Los podocitos son extremadamente sensibles al «shear stress» generado por el incremento de la presión de filtración a través de las hendiduras y sobre su superficie apical31,32.
La GEFS maladaptativa surge de los procesos descritos anteriormente, que implican un aumento de la tasa de filtración glomerular de cada nefrona, lo que lleva a un círculo vicioso de hipertrofia glomerular, hipertrofia podocitaria, estrés podocitario y su depleción con la eventual formación de sinequias y un exceso de deposición de matriz extracelular dentro del glomérulo31,33.
Las infecciones virales pueden inducir GEFS secundaria, ya sea por infección directa o por liberación de citoquinas inflamatorias que interactúan con los receptores de los podocitos. El VIH (virus de la inmunodeficiencia humana), parvovirus B-19, citomegalovirus, virus de Epstein-Barr, coronavirus-2 asociado a síndrome respiratorio agudo grave (SARS-CoV-2) o el virus del simio 40 (SV40) son algunos ejemplos de infecciones virales que pueden inducir estas lesiones1,34,35.
Existen algunos fármacos que pueden inducir GEFS, como el interferón -α, -β, o –γ, los bisfosfonatos intravenosos, las antraciclinas, el litio o los inhibidores de mTOR (mammalian target of rapamycin). Estas lesiones suelen ser reversibles una vez suspendido el tratamiento28,35.
No existe un patrón histológico típico en la GEFS secundaria, pero la glomerulomegalia es característica de aquellas formas adaptativas, la variante perihiliar (clasificación de Columbia) también suele ser característica en esta forma de GEFS en la que las sinequias están presentes predominantemente a nivel perihiliar35. Las formas colapsantes son más habituales en casos secundarios a VIH o fármacos como los bisfosfonatos. En cambio, en todas las formas de GEFS secundaria la fusión pedicelar es parcheada, ocupando generalmente <40% de la superficie glomerular. La presencia de fusión pedicelar difusa elimina el mecanismo de maladaptación como causa de GEFS primaria18,35.
La característica bioquímica que distingue a las formas secundarias es la presencia de albúmina sérica normal, a diferencia de la GEFS primaria que debuta con síndrome nefrótico. La presentación temporal de las manifestaciones clínicas puede tener también gran importancia para distinguir entre formas primarias y secundarias. Las formas primarias se suelen manifestar con un síndrome nefrótico de instauración brusca, mientras que las formas secundarias, en concreto las maladaptativas, a menudo cursan con proteinuria progresiva. Estas formas no responden a la terapia inmunosupresora y el tratamiento se debe dirigir, en la medida de lo posible, a resolver la causa subyacente. El manejo se basa en medidas que disminuyen la hiperfiltración glomerular, la retirada del fármaco causante o el tratamiento de la infección causante. Lograr una respuesta completa después del tratamiento con bloqueo del sistema renina-angiotensina (BSRAA), inhibidores del cotransportador sodio-glucosa tipo 2 (iSGLT2)36 o tratamiento de la causa subyacente, como la pérdida de peso en pacientes con obesidad, apoyaría el diagnóstico de GEFS secundaria18,37. Recientemente se ha publicado un ensayo clínico en pacientes con GEFS en los que el tratamiento con sparsertan (bloqueante dual de la endotelina tipo A y de la angiotensina II) redujo significativamente la proteinuria, sin que este efecto se tradujera en retrasar la progresión renal38. Aunque este estudio fue diseñado para analizar el efecto del sparsentan en pacientes con GEFS presuntamente primaria, el hecho de que la mediana de la proteinuria de los pacientes incluidos fue de rango subnefrótico (cociente proteínas-creatinina en orina de 3,1g/g) con una albúmina sérica media normal (3,49g/dl), indica que una amplia proporción de los pacientes estudiados no tenían GEFS primarias, sino que más bien se trataba de formas secundarias o hereditarias de GEFS, por lo tanto indicando su utilidad como antiproteinúrico en el manejo de dichos pacientes.
Glomeruloesclerosis segmentaria y focal secundaria de causa genéticaLa GEFS de causa genética o hereditaria es la menos frecuente, o quizás la menos diagnosticada. En los algoritmos actuales, el estudio genético no está establecido como parte del estudio inicial, por lo que en este documento queremos enfatizar que existen pacientes adultos en los cuales un estudio genético estaría indicado como primera evaluación ante la sospecha de GEFS, al igual que la biopsia renal.
Las técnicas de secuenciación masiva han permitido identificar más de 60 genes responsables de daño podocitario. Los genes que codifican proteínas localizadas en el podocito, diafragma de hendidura y la membrana basal glomerular son los principales implicados en la GEFS de causa genética, siendo los más frecuentes los genes del colágeno tipo IV, seguido de los genes relacionados con podocitopatías (ACTN4, INF2, NPHS1, NPHS2, TRPC6, LMXB1)8,39–41. Una de las indicaciones para realizar estudio genético en pacientes adultos es la no respuesta a inmunosupresión, aunque existen algunos casos de GEFS genética que podrían responder a corticoides (EMP2 o PLCG2)39 y algunas formas también son respondedoras a ICN (NPHS1, NPHS2, TRPC6, WT1) debido a un efecto local sobre la función de los podocitos y la estabilización de la sinaptopodina23, induciendo una reducción en la proteinuria, que puede ser etiquetada de remisión parcial42, pero en ningún caso de causa genética se alcanzará la remisión completa43,44.
La prevalencia de la GEFS de causa genética es elevada en niños (20-30%)39, la prevalencia en adultos varía desde alrededor del 22% en pacientes con historia familiar de enfermedad renal, hasta 10% en aquellos sin antecedentes familiares o en cohortes inespecíficas8,40,41,45,46. De ahí que el estudio genético se recomiende de forma general en pacientes con síndrome nefrótico de inicio temprano, especialmente en las formas resistentes a esteroides. Sin embargo, en población adulta la recomendación es más compleja. Las guías KDIGO recomiendan hacer una valoración individual de cada caso para indicar estudio genético, considerando como características a tener en cuenta la coexistencia de cuadros sindrómicos o de antecedentes familiares. La presencia de historia familiar aumenta la probabilidad de que una mutación sea la responsable del daño podocitario, si bien en una serie recientemente publicada hasta el 55% de los casos con variantes genéticas no tenían antecedentes familiares evidentes22. Esta misma publicación ha desvelado que en pacientes con proteinuria de lenta progresión sin una causa secundaria evidente, se encontró una variante genética en el 33% de los casos estudiados. Este porcentaje fue incluso mayor cuando el paciente presentaba microhematuria persistente con hematíes dismórficos, circunstancia que nos debe hacer sospechar una enfermedad relacionada con el colágeno IV.
El microscopio electrónico no resulta de utilidad para diagnosticar o sospechar GEFS de etiología genética. Aquellas formas con síndrome nefrótico completo en el momento de la biopsia renal mostrarán fusión pedicelar extensa, mientras que aquellas sin síndrome nefrótico mostrarán una fusión pedicelar solo parcial. Ocasionalmente, en GEFS asociada a mutaciones del colágeno IV, el microscopio electrónico muestra una membrana basal glomerular fina (<180nm en niños y <250nm en adultos)47, dato que podría ser indicación de estudio genético.
En la actualidad, el método más usado para el diagnóstico genético es la secuenciación masiva. Estas técnicas permiten la secuenciación simultánea de los exones de un conjunto de genes, o de todos los exones (secuenciación de exoma) o del genoma completo. La técnica de Sanger actualmente se utiliza para secuenciar genes pequeños o para confirmar una variante identificada previamente por secuenciación masiva48–50. Los paneles de genes asociados a varias enfermedades han resultado ser útiles en nefrología, por lo que son una buena herramienta cuando buscamos enfermedades sin un fenotipo específico, como en el caso de la GEFS de causa genética51,52.
Indicaciones de estudio genético en GEFSDe acuerdo con las situaciones de la práctica clínica habitual y a las dificultades que puede suponer en ciertos centros el acceso a la realización de estudios genéticos, hemos planteado el algoritmo de actuación mostrado en la figura 1.
.")
Aunque muchos de los genes causantes de GEFS dan lugar a una enfermedad con inicio en la infancia, el amplio espectro fenotípico no permite descartar estos genes como causante de una enfermedad de inicio en edad adulta. Por este motivo, los paneles de genes deben incluir todos los genes causantes de nefropatía glomerular.
Algunos genes causantes de otras enfermedades hereditarias, como tubulopatías (CLCN5, OCRL), microangiopatía trombótica (DKGE, CFH), ciliopatías (UMOD, NPHP4)53, enfermedad de depósito como la enfermedad de Fabry (GLA)39 y genes causantes de anomalías congénitas del riñón y tracto urinario (CAKUT pos sus siglas en inglés) podrían presentarse con un patrón de GEFS en la biopsia54.
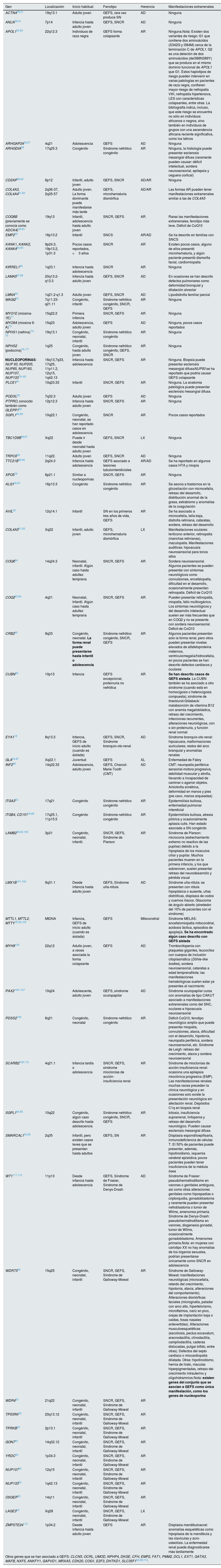
La tabla 1 describe los genes que se han asociado a GEFS de causa genética y sus características clínicas. Se han dividido de acuerdo a si los pacientes presentan manifestaciones extrarrenales como parte de un cuadro sindrómico o si son causantes de GEFS como única manifestación. Hasta la actualidad hay más de 60 genes asociados; sin embargo, tenemos que tener en cuenta que conforme avanza la investigación se van descubriendo nuevos genes, y que la GEFS puede deberse a alteraciones del podocito, de la membrana basal glomerular y sus componentes, pero también puede ser la cicatriz de enfermedad renal de cualquier origen54 (tabla 1).
Genes asociados a glomeruloesclerosis focal y segmentaria y sus características clínicas
| Gen | Localización | Inicio habitual | Fenotipo | Herencia | Manifestaciones extrarrenales |
|---|---|---|---|---|---|
| ACTN439,41 | 19q13.1 | Adulto joven | GEFS, rara vez produce SN | AD | Ninguna |
| ANLN39,41 | 7p14 | Infancia hasta adulto joven | GEFS, SNCR | AD | Ninguna |
| APOL155,56 | 22q12.3 | Individuos de raza negra | GEFS forma colapsante | AR | Ninguna.Nota: Existen dos variantes de riesgo: G1 que contiene dos aminoácidos (S342G y I384M) cerca de la terminación C de APOL1. G2 es una deleción de dos aminoácidos (del388N389Y) que se produce en el mismo dominio funcional de APOL1 que G1. Estos haplotipos de riesgo pueden intervenir en varias patologías en pacientes de raza negra, confieren mayor riesgo de nefropatía VIH, nefropatía hipertensiva, LES con características colapsantes, entre otras. La bibliografía indica, incluso, que este riesgo se encuentra no sólo en individuos africanos o negros, sino también en individuos de grupos con una ascendencia africana reciente significativa, como los latinos |
| ARHGAP2439,57 | 4q21 | Adolescencia | GEFS | AD | Ninguna |
| ARHGDIA58 | 17q25.3 | Congénito | Síndrome nefrótico congénito | AR | Ninguna, la histología puede presentar esclerosis mesangial difusa (raramente pueden causar: déficit intelectual, sordera neurosensorial, epilepsia y ceguera cortical) |
| CD2AP59,60 | 6p12 | Infantil, adulto joven | GEFS, SNCR | AD/AR | Ninguna |
| COL4A3, COL4A461,62 | 2q36-37, 2q35-57 | Adulto joven. La forma dominante puede manifestarse más tarde | GEFS, microhematuria dismórfica | AD/AR | Las formas AR pueden tener manifestaciones extrarrenales similar a las de COL4A5 |
| COQ8B (previamente se conocía como ADCK4)39,63 | 19q13 | Infantil, adolescencia hasta adulto joven | SNCR, GEFS | AR | Raras las manifestaciones extrarrenales, fenotipo más leve. Déficit de CoQ10 |
| EMP239 | 16p13.2 | Infantil | SNCS | AR/AD | Se ha descrito en familias con SNCS |
| KANK1, KANK2, KANK464,65 | 9p24.3, 19p13.2, 1p31.3 | Pocos casos reportados, <3 años | SNCR | AR | Existen pocos casos, alguno de ellos presentó microhematuria, y algún paciente presentó dismorfia facial, cardiomiopatía |
| KIRREL166 | 1q23.1 | Infancia hasta adolescencia | SNCR | AR | Ninguna |
| LAMA567,68 | 20q13.2-q13.3 | Infancia hasta adulto joven | GEFS, SNCR | AD | En ocasiones se han descrito defectos pulmonares como deformidad bronquial y dilatación alveolar |
| LMNA69 | 1q21.2-q1.3 | Adulto joven | GEFS, SNCR | AD | Lipodistrofia familiar parcial |
| MAGI270 | 7q11.23-q21.11 | Congénito, infantil | Síndrome nefrótico congénito, SNCR, GEFS | AR | Ninguna |
| MYO1E (miosina-1E)71 | 15q22.2 | Primera infancia | SNCR, GEFS | AR | Ninguna |
| MYO9A (miosina 9-A)72 | 15q23 | Adolescencia, adulto joven | GEFS | AD | Ninguna, pocos casos reportados |
| NPHS1 (nefrina)73–75 | 19q13.1 | Congénito, neonatal, infantil | Síndrome nefrótico congénito | AR | Ninguna |
| NPHS2 (podocina)75–77 | 1q25 | Congénito, hasta adulto joven | Síndrome nefrótico congénito, GEFS, SNCR | AR | Ninguna |
| NUCLEOPORINAS: NUP 93, NUP205, NUP85, NUP160, NUP107, NUP13378–80 | 16q13,7q33, 17q25, 11p11.2, 12q15, 1q42.13 | Infancia hasta adolescencia | SNCR, GEFS | AR | Ninguna. Biopsia puede presentar esclerosis mesangial difusaNUP93 se ha reportado que podría causar GEFS colapsante |
| PLCE181 | 10q23.33 | Infantil | SNCR, GEFS | AR | Ninguna. La anatomía patológica puede presentar esclerosis mesangial difusa |
| PODXL82 | 7q32.3 | Adulto joven | GEFS | AD | Ninguna |
| PTPRO, conocido también como GLEPP183 | 12p12.3 | Infancia hasta adulto joven | SNCR, GEFS | AR | Ninguna |
| SGPL184,85 | 10q22.1 | Congénito, neonatal, se han reportado casos en adolescencia | SNCR | AR | Pocos casos reportados |
| TBC1D8B86,87 | Xq22 | Puede ir desde neonatal hasta adulto joven | GEFS, SNCR | LX | Ninguna |
| TRPC639 | 11q22. | Adulto joven | GEFS, SNCR | AD | Ninguna |
| TTC21B88,89 | 2q24.3 | Infancia hasta adolescencia | GEFS asociado a lesiones tubulointersticiales | AR/AD | Se ha reportado en algunos casos HTA y miopía |
| XPO578 | 6p21.1 | Similar a nucleoporinas | SNCR, GEFS | AR | Ninguna |
| ALG190,91 | 16p13.3 | Congénito | Síndrome nefrótico congénito | AR | Se asocia a trastornos en la glicosilación con microcefalia, retraso del desarrollo, distribución anormal de la grasa, estrabismo y anomalías de la coagulación |
| AVIL92 | 12q14.1 | Infantil | SN en los primeros tres años de vida, GEFS | AR | Se ha asociado a microcefalia, talla baja, distrofia retiniana, cataratas, sordera, retraso del desarrollo |
| COL4A561,93 | Xq22 | Infantil, adulto joven | GEFS, microhematuria dismórfica | LX | Manifestaciones oculares: lenticono anterior, retinopatía (manchas retinianas), maculopatía. Manifestaciones auditivas: hipoacusia neurosensorial para tonos altos |
| COQ694 | 14q24.3 | Neonatal, infantil. Algún caso hasta adultez temprana | SNCR, GEFS | AR | Sordera neurosensorial. Algunos pacientes se pueden presentar con síntomas neurológicos como convulsiones, encefalopatía, dificultad en el desarrollo, ocasionalmente presentan retinopatía. Déficit de CoQ10 |
| COQ263,94 | 4q21 | Neonatal, Infantil. Algún caso hasta adultez temprana | SNCR, GEFS | AR | Pueden presentar retinopatía, miopatía, fallo multiorgánico. Los síntomas neurológicos y del desarrollo intelectual suelen ser más frecuentes que en COQ2 y no se presenta con sordera neurosensorial. Déficit de CoQ10 |
| CRB295 | 9q33 | Congénito, neonatal. La forma renal puede presentarse hasta infantil o adolescencia | Síndrome nefrótico congénito, SNCR, GEFS | AR | Algunos pacientes presentan solo la forma renal, pero otros pueden presentar niveles elevados de alfafetoproteína maternos, ventriculomegalia/hidrocefalia, en pocos pacientes se han descrito defectos cardíacos y oculares |
| CUBN96 | 10p13 | Infancia | GEFS excepcional, proteinuria no nefrótica | AR | Se han descrito casos de GEFS aislada. La CUBN también se ha asociado a otro síndrome (cuando está en homocigosis o heterocigosis compuesta); síndrome de Imerslund-Gräsbeck: malabsorción de vitamina B12 con anemia megaloblástica, retraso del crecimiento, infecciones recurrentes, alteraciones neurológicas, con o sin proteinuria, y función renal normal |
| EYA118 | 8q13.3 | Infancia, GEFS de inicio adulto (cuando es aislada) | GEFS, SNCR, Síndrome branquio-oto-renal | AD | Síndrome branquio-oto-renal: hipoacusia, malformaciones auriculares, restos del arco branquial y anomalías renales |
| GLA54,97 | Xq22.1 | Juventud | GEFS | XL | Enfermedad de Fabry |
| INF298 | 14q32.33 | Adolescencia, adulto joven | GEFS, Charcot-Marie-Tooth (CMT) | AD | CMT: neuropatía periférica sensorial-motora progresiva, debilidad muscular y atrofia, llevando a incapacidad de caminar o agarrar objetos. Amiotrofia simétrica, deformidad en manos y pies (pie cavo, manos arqueadas) |
| ITGA393 | 17q21 | Congénito | Síndrome nefrótico congénito | AR | Epidermólisis bullosa, enfermedad pulmonar intersticial |
| ITGB4, CD15154,99 | 17q25.1, 11p15.5 | Congénito | Síndrome nefrótico congénito | AR | Epidermólisis bullosa, atresia pilórica y ocasionalmente aplasia cutis. Han estado asociada a SN congénito |
| LAMB239,93,100 | 3p21 | Congénito, neonatal, infantil. | SNCR, GEFS, Síndrome de Pierson | AR | Síndrome de Pierson: microcoria (estrechamiento extremo no reactivo de las pupilas) debido a la hipoplasia de los músculos ciliar y pupilar. Muchos pacientes mueren en la primera infancia, y los que sobreviven, suelen presentar retraso del neurodesarrollo y pérdida visual |
| LMX1B101,102 | 9q31.1 | Desde infancia hasta adulto joven | GEFS, Síndrome uña-rótula | AD | Síndrome uña-rótula: se presentan con rótula hipoplásica o ausente, uñas distróficas, displasia de codos y cuernos ilíacos. Glaucoma de ángulo abierto (alrededor del 10% de pacientes con el síndrome) |
| MTTL1, MTTL2, MTTY18,39,103 | MtDNA | Infancia, GEFS de inicio adulto (cuando es aislada) | GEFS | Mitocondrial | Síndrome MELAS: encefalomiopatía mitocondrial, acidosis láctica, episodios de apoplejía. Se ha encontrado algún caso descrito con GEFS aislada |
| MYH9104 | 22q12 | Adulto joven, a veces asociada la forma colapsante | GEFS | AD | Trombocitopenia con plaquetas gigantes, leucocitos con cuerpos de inclusión citoplasmática (Döhle-like bodies), sordera neurosensorial, cataratas a edad tempranaNota: las manifestaciones hematológicas suelen estar ya presentes al nacimiento |
| PAX2105–107 | 10q24 | Adolescente, adulto joven | GEFS, síndrome oculopapilar | AD | Síndrome oculopapilar cursa con anomalías de tipo CAKUT asociado a manifestaciones extrarrenales como del SNC, oculares e hipoacusia neurosensorial |
| PDSS2108 | 6q21 | Congénito, neonatal | Síndrome nefrótico congénito | AR | Déficit CoQ10, fenotipo neurológico amplio que puede presentar miopatía, convulsiones, ataxia, dificultad con el desarrollo, hipotonía, neuropatía periférica, sordera neurosensorial, etc. Síndrome de Leigh: retraso del crecimiento, ataxia y sordera neurosensorial |
| SCARB2109,110 | 4q21.1 | Infancia tardía o adolescencia | SNCR, GEFS, síndrome mioclonías de acción-insuficiencia renal | AR | Síndrome de mioclonías de acción-insuficiencia renal: ocasiona una epilepsia mioclónica progresiva (EMP). Las manifestaciones renales muchas veces preceden la clínica neurológica y en ocasiones solo existe la presentación neurológica sin afectación renal. Depósitos C1q en biopsia renal |
| SGPL184,85 | 10q22 | Congénito, algún caso descrito hasta adolescencia | Síndrome nefrótico congénito, SNCR, GEFS | AR | Ictiosis, insuficiencia suprarrenal, linfopenia y retraso del desarrollo neurológico. Pueden causar esclerosis mesangial difusa |
| SMARCAL154,95 | 2q35 | Infantil, pero existen casos leves que se presentan hasta adultos | GEFS, SN | AR | Displasia espondiloepifisaria, inmunodeficiencia de células T. El 50% de pacientes puede presentar, además, hipotiroidismo, isquemia cerebral episódica; pocos pacientes pueden tener insuficiencia de la médula ósea |
| WT1111,112 | 11p13 | Desde infancia hasta adolescencia | GEFS, Síndrome de Frasier, Síndrome de Denys-Drash | AD | Síndrome de Frasier: pseudohermafroditismo en varones o genitales ambiguos, así como otras alteraciones genitales como hipospadias o criptorquidia, gonadoblastoma y raramente pueden presentar nefroblastoma o tumor de Wilms, amenorrea primaria. Síndrome de Denys-Drash: pseudohermafroditismo en varones, disgenesia gonadal, tumor de Wilms, ocasionalmente gonadoblastoma. Amenorrea primaria.Nota: en mujeres con cariotipo XX no hay anomalías de los órganos sexuales, podrían presentarse únicamente como SNCR en adolescencia |
| WDR7393 | 15q25 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | Síndrome de Galloway-Mowat: manifestaciones neurológicas (microcefalia, retardo del crecimiento, hipotonía, ataxia, alteraciones del comportamiento). Alteraciones dismórficas faciales (micrognatia, paladar con arco alto, hipertelorismo, microftalmos, nariz en pico, orejas de implantación baja o caídas, fosas nasales antevertidas). Alteraciones musculoesqueléticas (escoliosis, pectus excavatum, aracnodactilia, clinodactilia, campilodactilia, caderas dislocadas, pulgar bífido, entre otras). Defectos del septo cardiaco o miocardiopatía dilatada. Otras: hipotiroidismo, hernia de hiato, maculas hiperpigmentadas, retraso del crecimiento intrauterino y oligohidramnios.Nota: existen genes del conjunto que se asocian a GEFS como única manifestación, como los genes de nucleoporina |
| WDR493 | 21q22 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| TP53RK93 | 20q13.12 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| TPRKB93 | 2p13.1 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| GON793 | 14q32.12 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| YRDC93 | 1p34.3 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| NUP10793 | 12q15 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| NUP13393 | 1q42.13 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| OSGEP93 | 14q11 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | AR | |
| LAGE393 | Xq28 | Congénito, neonatal, infantil | SNCR, GEFS, Síndrome de Galloway-Mowat | LX | |
| ZMPSTE24113 | 1p34.2 | Desde infancia hasta adulto joven | GEFS | AR | Displasia mandibuloacral: anomalías esqueléticas como hipoplasia de la mandíbula y las clavículas y acro-osteólisis. La enfermedad renal puede diagnosticarse más tardíamente |
| Otros genes que se han asociado a GEFS: CLCN5, OCRL, UMOD, NPHP4, DKGE, CFH, EMP2, FAT1, PMM2, DCL1, EXT1, GATA3, MAFB, NXF5, ANKFY1, GAPVD1, MRXA5, CDK20, COG1, E2F3, DHTKD1, SLC35F154,99,114. | |||||
SN: síndrome nefrótico; SNCR: síndrome nefrótico corticorresistente; SNCS: síndrome nefrótico corticosensible; GEFS: glomeruloesclerosis focal y segmentaria; AR: autosómica recesiva; AD: autosómica dominante; LX: ligado al X; mtDNA: DNA mitocondrial.
Colores: Verde: GEFS como única manifestación; Naranja: GEFS como parte de cuadro sindrómico o con manifestaciones extrarrenales.
En la tabla 2 resumimos las indicaciones de estudio genético en pacientes con GEFS.
Indicaciones de estudio genético en adultos con glomeruloesclerosis focal y segmentaria
| Indicaciones de estudio genético en adultos con GEFS |
| 1. Pacientes con GEFS diagnosticada a cualquier edad y antecedentes familiares de ERC |
| 2. Pacientes con GEFS y cuadros sindrómicos |
| 3. Pacientes con GEFS y antecedentes familiares de consanguinidad |
| 4. Pacientes con GEFS diagnosticada a cualquier edad y presencia de microhematuria persistente con hematíes dismórficos |
| 5. Pacientes con GEFS y síndrome nefrótico corticorresistente |
| 6. Pacientes con GEFS y proteinuria de cualquier rango con albúmina normal, una vez descartada causa secundaria |
Los autores declaran no tener ningún conflicto de intereses.



