



Focal segmental glomerulosclerosis (FSGS) is a histological pattern of injury that derives from various pathological processes that affect podocytes, resulting in loss of selectivity of the glomerular filtration membrane, proteinuria and the development of renal failure that progresses to end-stage kidney disease in a significant number of patients. The classification proposed by the 2021 KDIGO guidelines divides FSGS into four categories: primary, secondary, genetic, and FSGS of undetermined cause, thus facilitating its diagnosis and management. Genetic causes of FSGS present significant clinical variability, complicating their identification. Genetic testing is crucial to identify FSGS of genetic cause. The prevalence of genetic FSGS is significant in children and considerable in adults, highlighting the importance of early diagnosis to avoid unnecessary treatments and facilitate genetic counselling. Massive sequencing techniques have revolutionized genetic diagnosis, allowing the identification of more than 60 genes responsible for podocyte damage. This document proposes clinical recommendations for carrying out genetic studies in adults with FSGS, highlighting the need for a correct classification for adequate therapeutic planning and improvement of results in clinical trials.
La glomeruloesclerosis focal y segmentaria (GEFS) es un patrón histológico de lesión que deriva de diversos procesos patológicos que afectan a los podocitos, resultando en pérdida de selectividad del filtrado glomerular, proteinuria y desarrollando insuficiencia renal que progresa a enfermedad renal crónica terminal en un importante número de pacientes. La clasificación propuesta por las guías KDIGO, en 2021, divide la GEFS en cuatro categorías: primaria, secundaria, genética y de causa no determinada, facilitando así su diagnóstico y manejo. Las causas hereditarias de la GEFS presentan una variabilidad clínica significativa, complicando su identificación. El estudio genético es crucial para identificar la GEFS de causa genética. La prevalencia de la GEFS genética es significativa en niños y considerable en adultos, destacando la importancia del diagnóstico temprano para evitar tratamientos innecesarios y facilitar el consejo genético. Las técnicas de secuenciación masiva han revolucionado el diagnóstico genético, permitiendo la identificación de más de 60 genes responsables del daño podocitario. Este documento propone recomendaciones clínicas y patológicas para la realización de estudios genéticos en adultos con GEFS, subrayando la necesidad de una correcta clasificación para la planificación terapéutica adecuada y la mejora de los resultados en ensayos clínicos.
Focal segmental glomerulosclerosis (FSGS) should be understood as a histologic pattern, not as a disease. This histologic pattern is the result of a variety of pathologic processes that share a common damage on the podocytes; it involves the loss of glomerular filtrate selectivity and clinically it is manifested as proteinuria and long-term renal failure.1 Histologically, the loss of podocytes involves the migration of epithelial cells from the Bowman’s capsule to the glomeruli in an attempt to replace the damaged podocytes, however this attempt of regeneration is inefficient and produces patchy mesangial sclerosis of the glomerular tuft (segmental) that begins in some glomeruli but not all (focal).2 The KDIGO (Kidney Disease: Improving Global Outcomes) guidelines for the management of glomerular diseases published in 2021 propose a new etiopathogenic classification, and divided FSGS into four categories, which facilitates the diagnosis and therapeutic approach. Thus there should be different forms: primary, secondary, genetic and of undetermined cause.3 Family and personal history and the clinical presentation are not always sufficient to exclude a hereditary cause of the disease. The clinical presentation of FSGS with a genetic cause is extremely variable; with differences in age of presentation, degree of proteinuria, and progression of chronic kidney disease (CKD).
Initially the genetic forms were described as an onset during childhood, mainly associated with corticosteroid-resistant nephrotic syndrome. However, depending on the selection criteria, up to 30% of adult cases of FSGS may be associated with a genetic cause,4–8 so it remains a challenge to define the criteria to perform a genetic study. The clinical and histologic features that appear to better predict a genetic etiology are: the absence of response to immunosuppressive therapies, the presence of microhematuria, the absence of diffuse pedicellar fusion in the renal biopsy, and maintaining a normal serum albumin despite developing nephrotic range proteinuria6,9,10 although these last two characteristics may be modified depending on the time of diagnosis.
It is well established that recognizing the diagnosis of genetically caused FSGS is of vital importance for patients. It is clear that, early diagnosis avoids certain diagnostic tests such as scans and exposure to unnecessary immunosuppressive treatment, and also allows the diagnosis of asymptomatic carriers; screening for associated pathologies and facilitates genetic counseling. Unlike the primary forms of FSGS, recurrence of FSGS of genetic cause in renal transplantation is not a problem; but it is relevant in the development of other pathologies such as anti-glomerular basement membrane antibodies which occurs in 2%–3% of renal transplant patients with X-linked Alport syndrome.11 Finally, it also allows an adequate selection of potential living related kidney donors.
A correct classification of patients according to their clinical and histological characteristics is essential for decision making and the planning of a suitable decisions and the planning of an adequate therapeutic scheme. Unfortunately, many clinical trials have failed to because they included patients with different forms of FSGS without a correct stratification.
In the present document we propose recommendations, based on clinical and anatomopathological criteria, for the performance of a genetic study in adult patients with FSGS.
Primary focal and segmental glomerulosclerosisThe etiology of FSGS of immunologic etiology, classically called “primary FSGS”; is not yet fully elucidated. In recent decades it has been assumed that it is caused by a circulating permeability factor, which is speculated to consist of a group of cytokines that abruptly alter podocyte function, increasing the permeability of the glomerular filtration membrane. This group of cytokines has not yet been defined, and there are several molecules that have been proposed as possible causes of the disease. These include cardiotropic-like cytokine factor-1 (CLCF-1), soluble urokinase-type plasminogen activator receptor (suPAR), the anti-CD40 antibody, apolipoprotein A1 and the soluble form of calcium/calmodulin-serine protein kinase (CASK).12 In recent years it has been identified that a proportion of patients with minimal change disease are caused by nephrin antibody13 and more recently it has been confirmed its role in in primary FSGS especially in relapses after transplant.14 However, recently it has been found that only 9% of patients with primary FSGS are caused by these antibodies,15 so it remains to be clarified which are the agents involved in this permeability factor that contributes to the remaining cases of FSGS.
Clinically, primary FSGS is presented abruptly in the form of complete nephrotic syndrome (proteinuria ≥3.5 g/day, hypoalbuminemia, hypercholesterolemia and edema). Histologically, under the light microscope there are no specific features that would distinguish it from the other other forms of FSGS; by electron microscopy it is highlighted the presence of a diffuse pedicellar fusion that occupies more than 80% of the surface of the glomerular membrane which differentiates it from the secondary forms, and it is a determinant finding. In a series of adult and predominantly white patients with primary FSGS, the median pedicellar fusion was 100%,16 however it must be taken into account that several hereditary forms of FSGS can also present with a diffuse pedicellar fusion.
A morphometric analysis of pedicle width that excluded patients with hereditary forms of FSGS, found wider pedicels in patients with primary FSGS compared to those with secondary FSGS. A pedicellar width >1500 nm adequately differentiated primary from secondary FSGS.17 Histologic findings such as podocyte vacuolization and microvillous transformation are related to the amount of proteinuria, and although nonspecific, it is more frequently seen in primary than in secondary forms.18
The treatment of primary FSGS is immunosuppression, and due to its poor prognosis observed in the absence of treatment, early initiation of immunosuppressive therapy is recommended in cases of FSGS that present with complete nephrotic syndrome.19
Treatment with glucocorticoids is the current cornerstone of the management of primary FSGS.3 However, this recommendation is based on observational studies and there are no studies that have compared prednisone with placebo in the treatment of primary FSGS.20 A small clinical trial found that the association of mycophenolate with lower doses of steroids achieved similar results with a lower cumulative steroid dose.21
The response to corticosteroid therapy is a characteristic feature of primary FSGS, which would rule out other causes of FSGS, although the definition of glucocorticoid resistance is sometimes difficult. Recent studies have even demonstrate a genetic variant in up to 42% of patients with glucocorticoid resistance,22 so that the genetic study would be particularly justified in this group of patients to avoid unnecessary treatment with immunosuppressants.
Calcineurin inhibitors (CNI) are the standard treatment for patients with contraindications, intolerance, or resistance to glucocorticoids.3 The mechanism of action of glucocorticoids and CNIs in primary FSGS is not entirely clear; presumably these therapies interfere with the cellular sites of the putative permeability factors. However, the information is somewhat ambiguous as to whether the benefit of immunosuppressives depends solely on their immunosuppressive effect or whether it is due to a local effect on podocyte function and the stabilization of synaptopodin.23 The different series have shown in FSGS a response rate to glucocorticoids and/or CNI of 40%–70% of cases19; although this proportion is probably underestimated because of the inclusion of patients with secondary or hereditary forms; and probably the rate of response to appropriate immunosuppressive therapy is higher in patients correctly classified as primary FSGS.
Once genetic and secondary causes have been ruled out, the recurrence rate of primary FSGS in renal transplantation is up to 80% in first transplants.24 Early recurrence in transplantation is the hallmark of primary FSGS. The best treatment for recurrent FSGS in transplantation is not yet known. Prophylactic plasmapheresis does not reduce the risk of recurrence, although therapeutic plasmapheresis is considered a good first-line option as a therapy to eliminate permeability factors, especially in very early recurrences with severe proteinuria.25 Prophylactic treatment with rituximab has not been shown to be effective in reducing the risk of recurrence.26
Secondary segmental and focal glomerulosclerosisMost secondary forms of FSGS are due to a mismatch between glomerular load and glomerular capacity. The pathogenic mechanism is always common and consists of damage and adaptive podocyte loss secondary to increased demand on the glomerular demand (inability to adapt to glomerular size, ischemia due to hypoperfusion). This may occur due to circumstances that reduce renal mass, such as low nephron mass at birth, reflux nephropathy, renal dysplasia, or situations in which there is an increase in the glomerular filtration rate that exceeds the glomerular capacity; as in obesity, high protein intake or androgen abuse.27–29 Any chronic glomerular or tubular disease can reduce total nephron function and lead to maladaptive FSGS that overlaps with the primary disorder.30
Hyperfiltration and hypertension in the glomerular capillary represent the main mechanism of strain on the podocyte. Podocytes are extremely sensitive to the “shear stress” generated by increased filtration pressure through the clefts and on their apical surface.31,32
Maladaptive FSGS arises from the processes described above that involve an increase in the glomerular filtration rate of each nephron, leading to a vicious cycle of glomerular hypertrophy, podocyte hypertrophy, podocyte stress and glomerular filtration rate of each nephron, leading to a vicious circle of glomerular hypertrophy, podocyte hypertrophy, podocyte stress and depletion and its depletion with the eventual formation of synechiae and an excess deposition of extracellular matrix within the glomerulus.31,33
Viral infections can induce secondary FSGS, either directly or by release of inflammatory cytokines that interact with podocyte receptors. HIV (human immunodeficiency virus), parvovirus B-19, cytomegalovirus, Epstein–Barr virus, severe acute respiratory syndrome-associated coronavirus-2 (SARS-CoV-2) or simian virus 40 (SV40) are some examples of viral infections that can induce these lesions.1,34,35
There are some drugs that can induce FSGS, such as interferon-α, -β, or -γ, intravenous bisphosphonates, the anthracyclines, lithium, or mTOR (mammalian target of rapamycin) inhibitors. These lesions are usually reversible after discontinuation of treatment.28,35
There is no typical histologic pattern in secondary FSGS, but glomerulomegaly is characteristic of adaptive forms, the perihilar variant (Columbia classification) is also characteristic in this form of FSGS in which synechiae are predominantly present at the perihilar level.35 Collapsing forms are more common incases secondary to HIV or drugs such as bisphosphonates. In contrast, in all forms of secondary FSGS, the pedicellar fusion is patchy usually occupying <40% of the glomerular surface. The presence of diffuse pedicellar fusion exclude the maladaptive mechanism as a cause of primary FSGS.18,35
The biochemical characteristic that differentiates the secondary forms is the presence of normal serum albumin, unlike primary FSGS that debuts with nephrotic syndrome. The temporal presentation of clinical manifestations may also be of great also importance to distinguish between primary and secondary forms. Primary forms usually manifest with an abrupt onset nephrotic syndrome, whereas secondary forms, in particular maladaptive forms, often present with progressive proteinuria. These forms do not respond to immunosuppressive therapy and treatment should be directed, as far as possible, to resolve the underlying cause. Management is based on procedures that decrease glomerular hyperfiltration, withdrawal of the causative drug or treatment of the related infection. Achieving a complete response after treatment with blockade of the renin-angiotensin system (BSRAA), sodium-glucose cotransporter type 2 (iSGLT2) inhibitors36 or treatment of the underlying cause such as weight loss in patients with obesity, would support the diagnosis of secondary FSGS.18,37 Recently it has been published, a clinical trial in patients with FSGS in whom treatment with sparsertan (a dual endothelin A and angiotensin II blocker) significantly reduced proteinuria, but without delaying renal progression.38
Although this study was designed to study the effect of sparsentan in patients with presumably primary FSGS, the fact is the median proteinuria of the patients included was in the sub-nephrotic range (urine protein-to-creatinine ratio of 3.1 g/g with a serum albumin within the normal range of (3.49 g/g); indicating that a large proportion of the patients studied did not have primary FSGS but rather secondary or hereditary forms of FSGS, thus indicating its usefulness as an antiproteinuric in the management of these patients.
Segmental and focal glomerulosclerosis secondary to genetic causesFSGS of genetic or hereditary cause is the least frequent, or perhaps the least diagnosed. In current algorithms, a genetic study is not established as part of the initial workup, so in this manuscript we want to emphasize that there are adult patients in whom a genetic study would be indicated as a first evaluation before suspicion of FSGS as well as renal biopsy.
Massive sequencing techniques have made it possible to identify more than 60 genes responsible for podocyte damage. The genes encoding proteins located in the podocyte, slit diaphragm, and glomerular basement membrane are the most important involved in genetically caused FSGS, the most frequent being the type IV collagen genes followed by the genes related to podocytopathies (ACTN4, INF2, NPHS1, NPHS2, TRPC6, LMXB1).8,39–41 One of the indications for genetic study in adult patients is non-response to immunosuppression, although there are some cases of genetic FSGS that might respond to corticosteroids (EMP2 or PLCG2)39 and some forms alsorespond to CNI (NPHS1, NPHS2, TRPC6, WT1) due to a local effect on the function of podocytes and the stabilization of synaptopodin,23 inducing a reduction in proteinuria, which can be labeled as partial remission,42 but in in genetic forms a complete remission will not be achieved.43,44
The prevalence of genetically caused FSGS is high in children (20%–30%),39 the prevalence in adults ranges from around 22% in patients with a family history of kidney disease to 10% in those with no family history or in undefined cohorts.8,40,45,46,41 Hence, genetic testing is generally recommended in patients with early-onset nephrotic syndrome, especially in steroid-resistant forms. However, in the population adult population, the recommendation is more complex. The KDIGO guidelines recommend making an individual assessment of each case to indicate genetic study, considering as characteristics to be taken into account the coexistence of syndromic conditions or family history. The presence of a family history increases the probability that a mutation is responsible for the podocyte damage; although in recently published series, up to 55% of the cases with genetic variants had no evidence of family history.22 This same publication has revealed that in patients with slowly progressive proteinuria without an obvious secondary cause, a genetic variant was found in 33% of the cases studied. This percentage was even higher when the patient had persistent microhematuria with dysmorphic red blood cells, a circumstance that should lead us to suspect a disease related to collagen IV.
Electron microscopy is not useful for diagnosing or suspecting FSGS of genetic etiology. Those forms with complete nephrotic syndrome at the time of renal biopsy will show extensive pedicellar fusion while those without nephrotic syndrome will show only partial pedicellar fusion. Occasionally, in FSGS associated with collagen IV mutations the electron microscopy shows a thin glomerular basement membrane (<180 nm in children and <250 nm in adults),47 which could be an indication for genetic study.
Currently the most widely used method for genetic diagnosis is massive sequencing. These techniques allow the simultaneous sequencing of the exons of a set of genes, or of all the exons (exome sequencing) or of the entire genome. The Sanger technique is currently used to sequence small genes or to confirm a variant previously identified by mass sequencing.48–50 Gene panels associated with various diseases have proven to be useful in nephrology, so they are a good tool when we are looking for diseases without a specific phenotype as in the case of FSGS of genetic cause.51,52
Indications for genetic study in GEFSIn accordance with the situations of routine clinical practice and the difficulties that may arise in certain centers regarding the access to genetic studies, we have proposed the following algorithm: (Fig. 1).
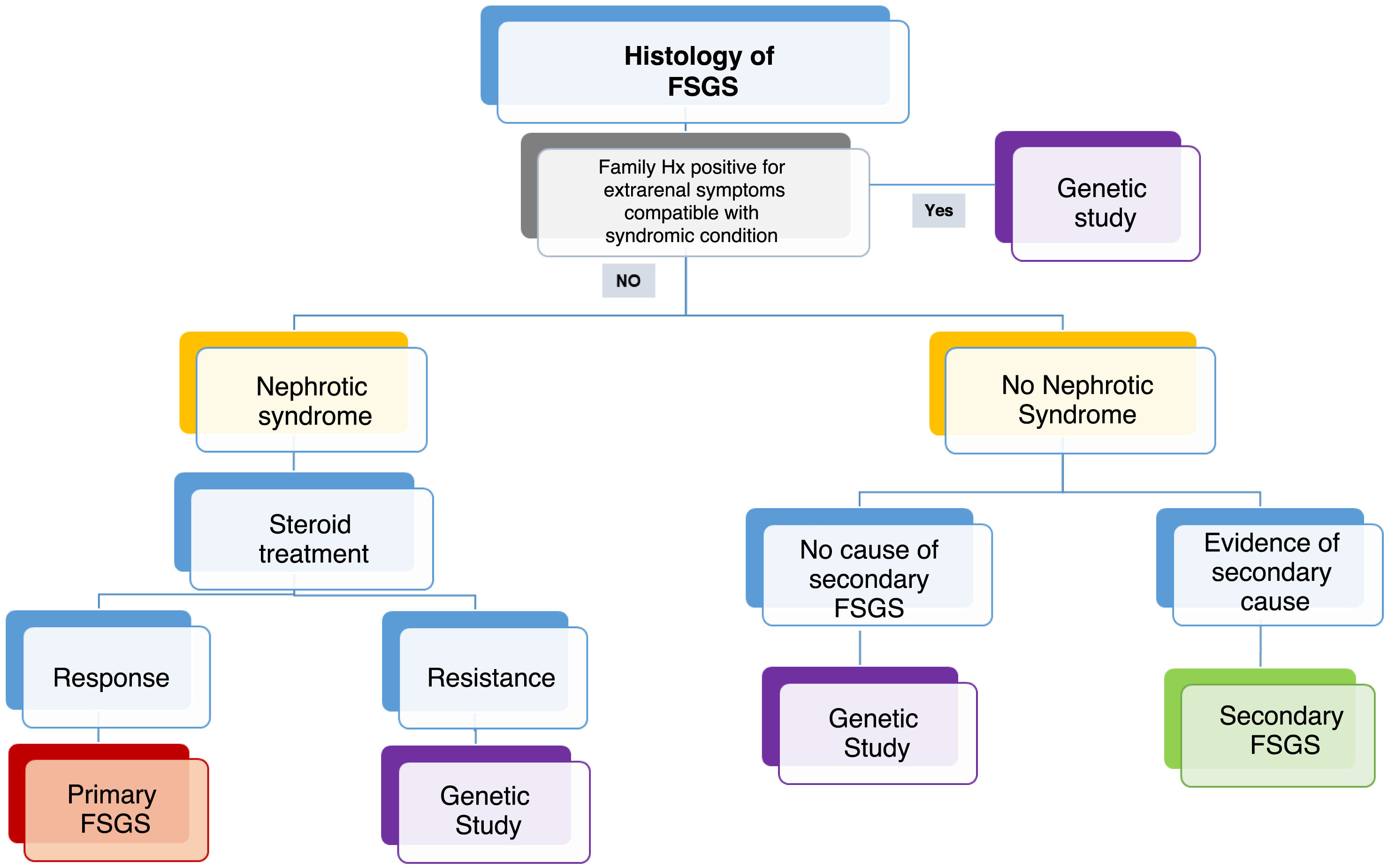
Although many of the genes that cause FSGS give rise to a disease with onset in childhood, the wide phenotypic spectrum does not allow us to rule out these genes as the cause of adult-onset disease. For this reason, gene panels should include all genes causing glomerular nephropathy.
Some genes that are the cause for other inherited diseases such as tubulopathies (CLCN5, OCRL), thrombotic microangiopathy (DKGE, CFH), ciliopathies (UMOD, NPHP4),53 deposit disease such as Fabry disease (GLA)39 and CAKUT-causing genes may present a FSGS pattern in the biopsy.54
Table 1 describes the genes that have been associated with FSGS of genetic cause and their clinical features. They have been divided according to whether patients present with extrarenal manifestations as part of a syndromic picture or whether they are the cause of FSGS as the only clinical expression. To date there are more than 60 associated genes; however, we have to take into account that as research progresses new genes are being discovered and FSGS may be due to alterations of the podocyte, the glomerular basement membrane and its components, but it may also be the scar of renal disease of any origin54 (Table 1).
Genes associated with focal and segmental glomerulosclerosis and their clinical characteristics.
| Gene | Location | Usual onset | Phenotype | Inheritance | Extra-renal manifestations |
|---|---|---|---|---|---|
| ACTN439,41 | 19q13.1 | Young adult | FSGS, rarely produces NS | AD | None |
| ANLN39,41 | 7p14 | Childhood to young adult | FSGS, NSCR | AD | None |
| APOL155,56 | 22q12.3 | Black individuals | FSGS collapsing collapsing form | AR | Note: there are two risk variants: G1 containing two amino acids (S342G and I384M) near the C-terminus of APOL1. The C terminus of APOL1. G2 is a deletion of two amino acids (del388N389Y) occurring in the same functional domain of APOL1 as G1. These risk haplotypes may be involved in several pathologies in black patients, conferring an increased risk of HIV nephropathy, hypertensive nephropathy, SLE with collapsing features, among others. The bibliography even indicates that this risk is found not only in African or black individuals, but also in individuals from groups with significant recent African ancestry, such as Latinos |
| ARHGAP2439,57 | 4q21 | Adolescence | FSGS | AD | None |
| ARHGDIA58 | 17q25.3 | Congenital | Congenital nephrotic syndrome | AR | None, the histology may show diffuse mesangial sclerosis (rarely may cause: intellectual deficit, sensorineural deafness, epilepsy and cortical blindness) |
| CD2AP59,60 | 6p12 | Childhood, young adult | FSGS, NSCR | AD/AR | None |
| COL4A3 COL4A461,62 | 2q36-37, 2q35-57 | Young adult the dominant form may manifest itself later in life | FSGS, microhematuria dysmorphic | AD/AR | |
| COQ8B (previously known as ADCK4)39,63 | 19q13 | Childhood, adolescence to young adult | NSCR, FSGS | AR | Rare extrarenal manifestations, milder phenotype. CoQ10 deficiecy |
| EMP239 | 16p13.2 | Childhood | NSCS | AR/AD | Described in families with NSCS |
| KANK1, KANK2, KANK464,65. | 9p24.3, 19p13.2,1p31.3 | Few cases reported, <3 years | NSCR | AR | Few cases, some with microhematuria, and some with facial dysmorphia, cardiomyopathy |
| KIRREL166 | 1q23.1 | Childhood until adolescence | NSCR | AR | None |
| LAMA567,68 | 20q13.2-q13.3 | Childhood to young adulthood | FSGS, NSCR | AD | Lung defects such as bronchial deformity and alveolar dilatation have been described occasionally |
| LMNA69 | 1q21.2-q1.3 | Young adulthood | FSGS, NSCR | AD | Familial partial lipodystrophy |
| MAGI270 | 7q11.23-q21.11 | Congenital, childhood | Congenital nephrotic syndrome, NSCR FSGS | AR | None |
| MYO1E (myosin-1E)71 | 15q22.2 | Early childhood | NSCR, FSGS | AR | None |
| MYO9A (myosin 9-A)72 | 15q23 | Adolescence, young adulthood | FSGS | AD | None, few cases reported |
| NPHS1 (nephrin)73–75 | 19q13.1 | Congenital, neonatal, childhood | Congenital nephrotic syndrome | AR | None |
| NPHS2 (podocin)75–77 | 1q25 | Congenital, through young adult | Congenital nephrotic syndrome, FSGS, NSCR | AR | None |
| NUCLEOPORINS:NUP 93, NUP205, NUP85, NUP160, NUP107, NUP13378–80 | 16q13,7q33, 17q25, 11p11.2, 12q15, 1q42.13 | Childhood through adolescence | NSCR, FSGS | AR | None. Biopsy may show diffuse mesangial sclerosis NUP93 has been reported to cause collapsing FSGS |
| PLCE181 | 10q23.33 | Childhood | NSCR, FSGS | AR | None. Pathology may show diffuse mesangial sclerosis |
| PODXL82 | 7q32.3 | Young adult | FSGS | AD | None |
| PTPRO, also known as GLEPP183 | 12p12.3 | Childhood to young adulthood | NSCR FSGS | AR | None |
| SGPL184,85 | 10q22.1 | Congenital, neonatal, cases reported in adolescence | NSCR | AR | Few cases reported |
| TBC1D8B86,87 | Xq22 | May range from neonatal to young adulthood | FSGS, NSCR | LX | None |
| TRPC639 | 11q22 | Young adult | FSGS, NSCR | AD | None |
| TTC21B88,89 | 2q24.3 | Childhood to adolescence | FSGS associated with tubulointerstitial lesions | AR/AD | Hypertension and myopia have been reported in some cases |
| XPO578 | 6p21.1 | Similar to nucleoporins | NSCR, FSGS | AR | None |
| ALG190,91 | 16p13.3 | Congenital | Congenital nephrotic syndrome | AR | Associated with glycosylation disorders with microcephaly, developmental delay, abnormal fat distribution, strabismus, and coagulation abnormalities |
| AVIL92 | 12q14.1 | Infancy | NS in the first three years of life, FSGS | AR | It has been associated with microcephaly, short stature, retinal dystrophy, cataracts, deafness, developmental delay |
| COL4A561,93 | Xq22 | Infancy young adult | FSGS, dysmorphic microhematuria | LX | Ocular manifestations: anterior lenticonus, retinopathy (retinal spots), maculopathy. Auditory manifestations: sensorineural hearing loss for high tones |
| COQ694 | 14q24.3 | Neonatal, infancy some cases up to early adulthood | NSCR, FSGS | AR | Sensorineural deafness. Some patients may present with neurological symptoms such as seizures, encephalopathy, developmental difficulty, occasionally presenting retinopathy. CoQ10 deficiency |
| COQ263,94 | 4q21 | Neonatal, childhood. Some cases up to early adulthood | NSCR, FSGS | AR | May present retinopathy, myopathy, multi-organ failure. Neurological and intellectual development symptoms are usually more frequent than in COQ2 and do not present with neurosensory deafness. CoQ10 deficiency |
| CRB295 | 9q33 | Congenital, neonatal the renal form may present until childhood or adolescence | Congenital nephrotic syndrome, CNRS, FSGS | AR | Some patients present only the renal form, but others may present elevated levels of maternal alpha-fetoprotein, ventriculomegaly/hydrocephalus, in a few patients cardiac and eye defects have been described |
| CUBN96 | 10p13 | Childhood | Exceptional FSGS non-nephrotic proteinuria | AR | There have been described cases of isolated FSGS. CUBN has been also associated with another syndrome (when it is homozygous or compound heterozygous); Imerslund-Gräsbeck syndrome: vitamin B12 malabsorption with megaloblastic anemia, growth retardation, recurrent infections, neurological abnormalities, with or without proteinuria, and normal renal function |
| EYA118 | 8q13.3 | Childhood, adult-onset FSGS (when isolated) | FSGS, NSCR, Branchio-oto-renal syndrome | AD | Branchio-oto-renal syndrome, hearing loss auricular malformations, remnants of the branchial arch, and renal abnormalities |
| GLA54,97 | Xq22.1 | Youth | FSGS | XL | Fabry disease |
| INF298 | 14q32.33 | Adolescence, young adult | FSGS Charcot-Marie-Tooth (CMT) | AD | CMT: progressive peripheral sensory-motor neuropathy, muscle weakness, and atrophy, causing inability to walk or grasp objects. Symmetrical amyotrophy, deformity in hands and feet (cavus foot, bowed hands) |
| ITGA393 | 17q21 | Congenital | Congenital nephrotic syndrome | AR | Epidermolysis bullosa, interstitial lung disease |
| ITGB4, CD15154,99 | 17q25.1, 11p15.5 | Congenital | Congenital nephrotic syndrome | AR | Epidermolysis bullosa, pyloric atresia, and occasionally aplasia cutis. They have been associated to congenital NS |
| LAMB239,93,100 | 3p21 | Congenital, neonatal, childhood. | NSCR, FSGS, Pierson syndrome | AR | Pierson syndrome: microcoria (extreme nonreactive narrowing of the pupils) due to hypoplasia of the ciliary and pupillary muscles. Many patients die in early childhood, and those who survive often have neurodevelopmental delay and visual loss |
| LMX1B101,102 | 9q31.1 | Infancy to young adulthood | FSGS, nail -patella syndrome | AD | Nail-patella syndrome: presents with hypoplastic or absent patella, dystrophic nails, elbow and iliac horn dysplasia. Open angle glaucoma (about 10% of patients with the syndrome) |
| MTTL1, MTTL2, MTTY18,39,103 | MtDNA | Childhood, adult-onset FSGS (when isolated) | FSGS | Mitochondrial | MELAS syndrome: mitochondrial encephalomyopathy, lactic acidosis, stroke episodes. Some cases have been described with isolated FSGS |
| MYH9104 | 22q12 | Young adult, sometimes associated with collapsing form | FSGS | AD | Thrombocytopenia with giant platelets, leukocytes with cytoplasmic inclusion bodies (Döhle-like bodies), sensorineural deafness, cataracts at an early age |
| Note: hematological manifestations are usually already present at birth | |||||
| PAX2105–107 | 10q24 | Adolescent, young adult | FSGS, oculopapillary syndrome | AD | Oculopapillary syndrome presents with CAKUT-type abnormalities associated with extrarenal manifestations such as CNS, ocular and sensorineural hearing loss |
| PDSS2108 | 6q21 | Congenital, neonatal | Congenital nephrotic syndrome | AR | CoQ10 deficiency, broad neurological phenotype that may present myopathy, seizures, ataxia, developmental difficulties, hypotonia, peripheral neuropathy, sensorineural deafness, etc. Leigh syndrome: growth retardation, ataxia and sensorineural deafness |
| SCARB2109,110 | 4q21.1 | Late childhood or adolescence | NSCR, FSGS, renal-failure action myoclonus syndrome | AR | Renal action-failure myoclonus syndrome: causes progressive myoclonic epilepsy (PME). Renal manifestations often precede neurological symptoms and sometimes only neurological presentation exists without renal involvement. C1q deposits in renal biopsy |
| SGPL184,85 | 10q22 | Congenital, some cases described up to adolescence | Congenital nephrotic syndrome, NSCR, FSGS | AR | Ichthyosis, adrenal insufficiency, lymphopenia and neurological developmental delay. They can cause diffuse mesangial sclerosis |
| SMARCAL154,95 | 2q35 | Infancy, but there are mild cases that occur up to adulthood | FSGS, NS | AR | Spondyloepiphyseal dysplasia, T-cell immunodeficiency. 50% of patients may also present hypothyroidism, episodic cerebral ischemia; few patients may have bone marrow insufficiency |
| WT1111,112 | 11p13 | From childhood to adolescence | FSGS, Frasier syndrome, Denys-Drash syndrome | AD | Frasier syndrome: pseudohermaphroditism in males or ambiguous genitalia, as well as other genital abnormalities such as hypospadias or cryptorchidism, gonad blastoma and rarely they may present nephroblastoma or Wilms tumor, primary amenorrhea. Denys-Drash syndrome: pseudo hermaphroditism in males, gonadal dysgenesis, Wilms tumor, occasionally gonadoblastoma. Primary amenorrhea. In women with XX karyotype there are no abnormalities of the sexual organs, they could only appear as NSCR in adolescence |
| WDR7393 | 15q25 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| WDR493 | 21q22 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | Galloway-Mowat syndrome: neurological manifestations (microcephaly, growth retardation, hypotonia, ataxia, behavioral changes). Dysmorphic facial alterations (micrognathia, high-arched palate, hypertelorism, microphthalmos, pointed nose, low-set or drooping ears, anteverted nostrils). Musculoskeletal alterations (scoliosis, pectus excavatum, arachnodactyly, clinodactyly, campylodactyly, dislocated hips, bifid thumb, among others). Cardiac septal defects or dilated cardiomyopathy. Others: hypothyroidism, hiatal hernia, hyperpigmented macules, intrauterine growth retardation and oligohydramnios. Note: there are genes in the set that are associated with FSGS as the only manifestation, such as the nucleoporin genes |
| TP53RK93 | 20q13.12 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| TPRKB93 | 2p13.1 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| GON793 | 14q32.12 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| YRDC93 | 1p34.3 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| NUP10793 | 12q15 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| NUP13393 | 1q42.13 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| OSGEP93 | 14q11 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | AR | |
| LAGE393 | Xq28 | Congenital, neonatal, chilhood | NSCR, FSGS, Galloway-Mowat syndrome | LX | |
| ZMPSTE24113 | 1p34.2 | Infancy to young adult | FSGS | AR | Mandibuloacral dysplasia: skeletal abnormalities such as hypoplasia of the jaw and clavicles and acro-osteolysis. Kidney disease may be diagnosed later |
Other genes that have been associated with FSGS: CLCN5, OCRL, UMOD, NPHP4, DKGE, CFH, EMP2, FAT1, PMM2, DCL1, EXT1, GATA3, MAFB, NXF5, ANKFY1, GAPVD1, MRXA5, CDK20, COG1, E2F3, DHTKD1, SLC35F1.54,99,114
NS: nephrotic syndrome; NSCR: steroid-resistant nephrotic syndrome; NSCS: steroid-sensitive nephrotic syndrome; FSGS: focal segmental glomerulosclerosis; AR: autosomal recessive; AD: autosomal dominant; LX: X-linked; mtDNA: mitochondrial DNA.
Colors: green: FSGS as the only manifestation; orange: FSGS as part of a syndromic picture or with extrarenal manifestations.
In Table 2 we summarize the indications for genetic study in patients with FSGS.
Indications for genetic study in adults with focal segmental glomerulosclerosis.
| Indications for genetic study in adults with FSGS |
| 1. Patients with FSGS diagnosed at any age and family history of CKD |
| 2. Patients with FSGS and syndromic symptoms |
| 3. Patients with FSGS and family history of consanguinity |
| 4. Patients with FSGS diagnosed at any age and presence of persistent microhematuria with dysmorphic red blood cells |
| 5. Patients with FSGS and steroid-resistant nephrotic syndrome |
| 6. Patients with FSGS and proteinuria of any range with albúmina normal, una vez descartada causa secundaria |



