



Una de las principales causas de morbimortalidad en el paciente con enfermedad renal crónica es la cardiovascular. La inflamación y las alteraciones en el metabolismo óseo mineral son una condición patológica que conlleva aumento del riesgo cardiovascular en la enfermedad renal crónica. Los parámetros bioquímicos clásicos del metabolismo óseo mineral como fósforo, calcio, vitamina D y PTH tienen una implicación muy conocida en el riesgo cardiovascular. Los nuevos marcadores, FGF23 y klotho, también podrían estar implicados en la enfermedad cardiovascular.
Cardiovascular factors are one of the main causes of morbidity and mortality in patients with chronic kidney disease. Bone mineral metabolism disorders and inflammation are pathological conditions that involve increased cardiovascular risk in chronic kidney disease. The cardiovascular risk involvement of bone mineral metabolism classical biochemical parameters such as phosphorus, calcium, vitamin D and PTH is well known. The newest markers, FGF23 and klotho, could also be implicated in cardiovascular disease.
La enfermedad renal crónica (ERC) es un problema de salud pública que va en aumento, con una incidencia y una prevalencia cada vez mayores (10% de la población general)1,2. La principal causa de mortalidad en el paciente con ERC es la cardiovascular (CV)3, con un incremento del riesgo de hasta 20 veces el de la población general incluso en estadios iniciales4–6. Hasta un 80% de los pacientes con ERC presentan enfermedad CV asociada: hipertensión arterial (36%), cardiopatía isquémica (22-39%), fibrilación auricular (30%), valvulopatía (24%) e hipertrofia ventricular izquierda (HVI) (50-75% en estadios 3-4 de ERC)7,8.
Factores de riesgo cardiovascular en el paciente con enfermedad renal crónicaEl mayor RCV de los pacientes con ERC se explica por la elevada presencia de factores de riesgo clásicos y la superposición de factores específicos del estado urémico así como por el estado inflamatorio de la ERC; en el estadio 5 se añaden otros factores relacionados con la diálisis o el trasplante que provocan un exceso de calcificación vascular9,10 (fig. 1). La enfermedad óseo mineral relacionada con la ERC (EOM-ERC) tiene un papel crucial en la ERC. La EOM-ERC integra anomalías bioquímicas, esqueléticas y calcificaciones extraesqueléticas que se producen por las alteraciones del metabolismo mineral en la ERC secundarias a la pérdida progresiva de masa y función renal. Se manifiesta por una o por la combinación de las siguientes manifestaciones11:
- •
Anormalidades del calcio (Ca), fósforo (P), hormona paratiroidea (PTH) y vitamina D, klotho y factor de crecimiento fibroblástico 23 (FGF23).
- •
Alteraciones del remodelado, mineralización, volumen, crecimiento o fragilidad del esqueleto.
- •
Calcificaciones cardiovasculares o de otros tejidos blandos.

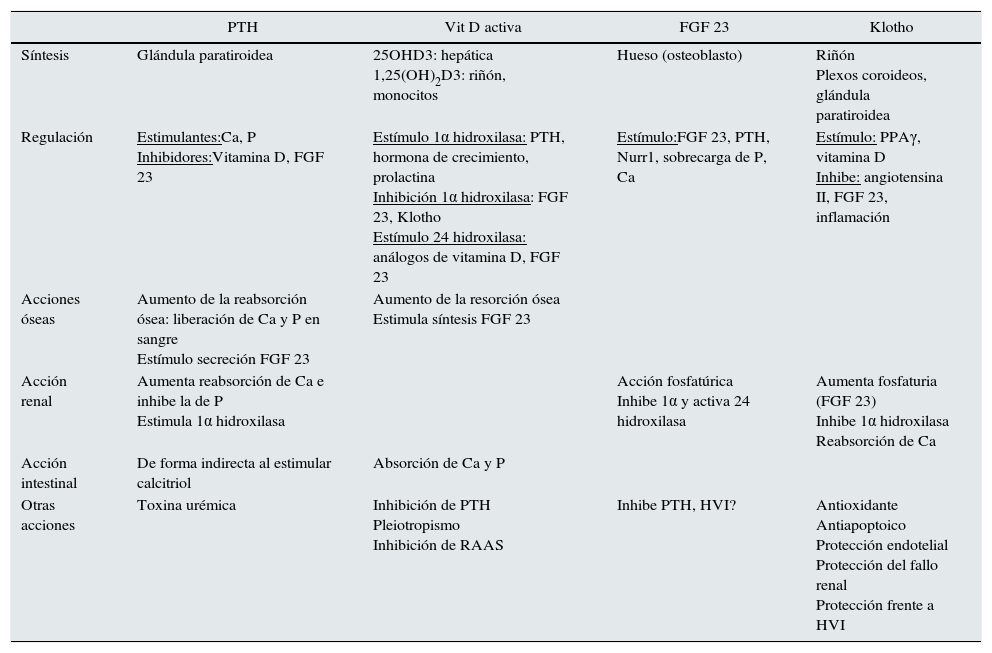
El estudio de los marcadores de EOM-ERC, en algunas ocasiones, nos permite predecir el RCV. En la tabla 1 resumimos el papel de los factores implicados en la EOM-ERC y a continuación revisaremos el papel de klotho y del FGF23 en la ECV.
Regulación de la enfermedad óseo mineral relacionada con la enfermedad renal crónica
| PTH | Vit D activa | FGF 23 | Klotho | |
|---|---|---|---|---|
| Síntesis | Glándula paratiroidea | 25OHD3: hepática 1,25(OH)2D3: riñón, monocitos | Hueso (osteoblasto) | Riñón Plexos coroideos, glándula paratiroidea |
| Regulación | Estimulantes:Ca, P Inhibidores:Vitamina D, FGF 23 | Estímulo 1α hidroxilasa: PTH, hormona de crecimiento, prolactina Inhibición 1α hidroxilasa: FGF 23, Klotho Estímulo 24 hidroxilasa: análogos de vitamina D, FGF 23 | Estímulo:FGF 23, PTH, Nurr1, sobrecarga de P, Ca | Estímulo: PPAγ, vitamina D Inhibe: angiotensina II, FGF 23, inflamación |
| Acciones óseas | Aumento de la reabsorción ósea: liberación de Ca y P en sangre Estímulo secreción FGF 23 | Aumento de la resorción ósea Estimula síntesis FGF 23 | ||
| Acción renal | Aumenta reabsorción de Ca e inhibe la de P Estimula 1α hidroxilasa | Acción fosfatúrica Inhibe 1α y activa 24 hidroxilasa | Aumenta fosfaturia (FGF 23) Inhibe 1α hidroxilasa Reabsorción de Ca | |
| Acción intestinal | De forma indirecta al estimular calcitriol | Absorción de Ca y P | ||
| Otras acciones | Toxina urémica | Inhibición de PTH Pleiotropismo Inhibición de RAAS | Inhibe PTH, HVI? | Antioxidante Antiapoptoico Protección endotelial Protección del fallo renal Protección frente a HVI |
Es una proteína de 251 aminoácidos de 32 kDa, sintetizada y secretada por las células óseas, principalmente el osteoblasto. Se incluye en el grupo de las hormonas «fosfatoninas»12 por su implicación en la eliminación renal de P. Ha sido relacionada fisiopatológicamente en los llamados «síndromes hipofosfatémicos raros»13, caracterizados por defecto en la mineralización y deformidades óseas, hipofosfatemia, pérdida renal de P y niveles inapropiadamente bajos de calcitriol. Es considerado uno de los principales factores en la regulación del metabolismo del P14–16. La acción biológica del FGF23 depende del gen klotho14,17–19 que actúa como su correceptor. El FGF23 también se expresa en el corazón, hígado, glándula tiroides y paratiroides, intestino y músculo esquelético19.
Regulación de FGF23La regulación del FGF23 viene determinada por:
- 1.
Vitamina D activa: el calcitriol aumenta la transcripción de FGF23 de manera directa y de manera indirecta mediante vías de señalización extracelulares mediadas por leptina e interleucina 620. El calcitriol también aumenta la expresión del receptor nuclear asociado a la proteína 1 (Nurr1) en células óseas y de PTH, lo que conlleva el aumento de FGF2321.
- 2.
Niveles de Ca: efecto estimulante de la secreción de FGF2322,23.
- 3.
Hiperparatiroidismo: el aumento de PTH relacionado con la ERC puede estimular la secreción de FGF23 a través de Nurr119,21. En el hiperparatiroidismo primario (HPTHP), hipotéticamente, FGF23 quedaría suprimido ya que la hipersecreción de PTH causaría hipofosfatemia y se suprimiría uno de los estímulos de secreción de FGF2324. Sin embargo, en estudios experimentales, se ha demostrado que ratones con HPTHP presentan niveles de FGF23 más elevados que los controles y que PTH es el factor estimulador25, ya que el FGF23 disminuye tras la paratiroidectomía25. Yamashita et al.24 demostraron en pacientes con HPTHP que los niveles de FGF23 estaban elevados frente a los controles sanos. No obstante, en este mismo estudio, los pacientes sin ERC ni HPTHP no presentaron diferencias en los niveles de FGF23 frente a los controles24, y concluyeron que en el HPTHP la función renal es determinante en los niveles de FGF2324.
- 4.
Niveles de P: los niveles séricos de P se correlacionan positivamente con las elevaciones de FGF23 en los pacientes con ERC26. Sin embargo, la restricción de P en la dieta de los pacientes con ERC muestra resultados contradictorios en el control de FGF23. Algunos estudios demuestran que la restricción de P en la dieta fracasa en descender los niveles de FGF23 en los pacientes con ERC estadio 3-427 y no los modifica en voluntarios sanos28. A pesar de ello, el descenso de la absorción de P con captores de fósforo, como sevelamero, disminuyen los niveles de FGF2329,30.
- 5.
Descenso de la síntesis renal de klotho: la afinidad de FGF23 por su receptor (especialmente FGFR1 a nivel renal) es muy baja31. En condiciones fisiológicas, el FGF23, al unirse al FGFR1, no sería capaz de generar transducción de señal31. Al agregar klotho, la afinidad del receptor aumenta significativamente y permite la activación de FGFR1 con concentraciones fisiológicas de FGF2331. El descenso de klotho podría causar una resistencia a la acción de FGF23; en la ERC esta resistencia conllevaría una reducción de la fracción de excreción de fosfato, y un aumento del P plasmático y, por tanto, secreción de FGF2332.
Como curiosidad, la infusión de hierro y los niveles bajos de hierro pueden inducir síntesis de FGF23, aunque no en su forma activa22. La acidosis metabólica, los estrógenos y la leptina también provocan aumentos de FGF2333,34.
Acciones biológicas de FGF23El FGF23 presenta receptores diana denominados FGFR 1, 3 y 4 y el receptor transmembrana β glucuronidasa. Para ejercer su acción sobre FGFR1 a nivel renal necesita de su correceptor klotho31,35.
- •
Hueso: en aquellos procesos caracterizados por defecto en la mineralización (raquitismo y osteomalacia) existe un exceso de producción o actividad biológica de FGF2336. Diversos grupos están investigando el efecto directo del FGF23 sobre el hueso, sin embargo, todavía no se ha demostrado evidencia de un efecto directo de FGF23 sobre el hueso37. En la regulación de la síntesis y la secreción del FGF23 han sido implicadas diversas proteínas expresadas predominantemente en el hueso como Phex (endopeptidasa reguladora del P ligada al cromosoma X) y glucoproteínas como la derivada de la matriz proteica de la dentina (DMP1) y de la matriz extracelular36,37. Determinadas alteraciones de estas proteínas producen un incremento de la actividad biológica por aumento de la expresión de FGF23, potenciando la fosfaturia y la hipofosfatemia, e inhibiendo la formación ósea36,37. Otras veces se puede producir el efecto contrario: un descenso de la actividad de FGF23 con incremento de los niveles de P sérico y calcificación anómala (calcicosis tumoral)38.
- •
Riñón: actúa sobre la homeostasis del P inhibiendo la expresión de los cotransportadores sodio-fosfato tipo ii (Na/P IIa y Na/P IIc), disminuyendo la reabsorción tubular de P a nivel de túbulo proximal e incrementando la excreción renal de P17,19,39. También disminuye los niveles de calcitriol, suprimiendo la actividad de la enzima 1α hidroxilasa (vía CYP27B1) y estimulando la enzima 24 hidroxilasa (vía CYP24A1)19,39,40. Por último, a nivel renal, inhibe la transcripción del gen klotho41.
- •
Paratiroides: el FGF23 disminuye la producción y secreción de PTH, esto ha sido demostrado por varios grupos de investigación: trabajos como el de Ben-Dov et al.42 y Krajisnk et al.43 indican que FGF23 produce una supresión de PTH in vivo e in vitro y disminuye la expresión-trascripción del ARNm y la secreción proteica de PTH43. No obstante, en glándulas paratiroideas urémicas hiperplásicas de rata, el FGF23 falla en la inhibición de la PTH frente a glándulas paratiroideas sanas; tal vez, debido a un descenso en la expresión de FGFR1 y klotho en las glándulas hiperplásicas urémicas44. El impacto de la paratiroidectomía, en pacientes con ERC, sobre los niveles de FGF23 fue estudiado por Takahashi et al.45, que diseñaron un estudio en 30 pacientes en hemodiálisis tratados mediante paratiroidectomía, con implante en antebrazo, y determinaron niveles de FGF23 y klotho, y concluyeron que FGF23 descendía y que klotho presentaba un descenso inicial con posterior elevación con respecto a los valores posparatiroidectomía.
- •
Corazón: en los miocardiocitos de ratones se ha demostrado que, a través de su receptor (FGFR-4), el FGF23 activa la vía de la calcineurina nuclear factor of activated T cells (NFAT) y provoca HVI de forma independiente de klotho46.
El paciente con ERC en estadios finales puede llegar a presentar valores de FGF23 de hasta 100 veces su valor normal28; además, los altos niveles de FGF23 predicen la progresión de ERC, como se ha ratificado en numerosos estudios47–49. Los niveles elevados de FGF23 se asocian a un aumento de la mortalidad ajustado para factores de riesgo clásico cardiovasculares y otros marcadores tradicionales de ERC47,50.
Se ha demostrado una asociación de FGF23 con la calcificación vascular, aunque no parece que FGF23 sea el inductor51. Scialla et al.51 estudiaron la asociación entre FGF23, P, calcificación coronaria y de la aorta torácica medida por TAC en 1.501 participantes con ERC (filtrado glomerular [FG] medio de 47±17ml/min/1,73 m2; estadios 2-4). Estos autores demostraron que FGF23 no se asociaba con la calcificación vascular medida por TAC y que, en estudios in vitro, el FGF23 no produce calcificación ni inducía calcificación en medios de cultivo con células del músculo liso (CMLV), ni presentaba expresión en la aorta de ratones y humanos51. El papel del FGF23 en la calcificación vascular vendría marcado por la hiperfosfatemia que sí induce calcificación vascular: las CMLV presentan una diferenciación osteoblástica en medios ricos en P52,53. Sí parece que hay asociación entre la gravedad de la calcificación y el FGF23 en la ERC, por lo que FGF23 podría ser un marcador de seguimiento y no de génesis de la calcificación vascular51.
El exceso de FGF23 conlleva un aumento de la morbimortalidad CV en la ERC54,55 de forma independiente del FG56, esto puede ser debido:
- 1.
A que el FGF23 reduce los niveles de vitamina D activa (calcitriol) al inhibir la 1α hidroxilasa y estimular la 24 hidroxilasa19,39.
- 2.
A que el FGF23 se relaciona con la presencia de marcadores inflamatorios57,58 y de estrés oxidativo como los productos de glucosilación avanzada que se asocian a calcificación vascular57.
- 3.
A que, en algunos estudios, el FGF23 se asocia con la proteinuria59.
- 4.
Al papel del FGF23 en la génesis de la HVI (importante generador de arritmias y de fallo cardíaco): el elegante estudio de Faul et al.46 evidenció, en una cohorte de más de 3.000 pacientes con ERC con FG entre 20-70ml/min, que había una correlación entre los niveles de FGF23 y la HVI. En este mismo estudio se detalló como FGF23 provocaba un aumento de α–actina en miocardiocitos de rata con un aumento de la expresión de marcadores de HVI, como β-miosina de cadena pesada fetal, y un descenso de la α-miosina de cadena pesada adulta. Este mecanismo es independiente de klotho y es mediado por la activación de la vía de la calcineurina NFAT.
Es una proteína transmembrana de 130 kDa que se expresa predominantemente en el riñón (túbulo distal, proximal y colector), en la glándula paratiroidea, plexo coroideo y también a nivel endotelial60,61. Presenta 3 formas diferentes62,63: klotho-cut de poca repercusión biológica, la forma completa, unida a la membrana (actúa como correceptor de FGF23), y la forma secretada.
Regulación de klotho- 1.
En modelos experimentales in vitro se ha demostrado que la señal peroxisome proliferator activated receptor γ (PPAR γ)64 aumenta la síntesis de klotho. El calcitriol también aumenta la expresión de klotho en modelos animales con ERC e ingesta elevada de P65.
- 2.
Los factores que disminuyen la síntesis de klotho son: el FGF2366–68 el estrés oxidativo69 y la angiotensina II (a través de sus receptores tipo I y del aumento de la enzima conversora del factor de necrosis tumoral α TACE)17,70,71. El gen klotho, al ser de síntesis predominantemente renal, está disminuido en los pacientes con ERC62,72.
- •
Riñón: el klotho induce fosfaturia directamente, actuando a nivel de túbulo proximal al inhibir el cotransportador Na/P IIa y Na/P IIc72–74. Además, klotho es correceptor de FGFR-1 y, por lo tanto, facilita la acción fosfatúrica de FGF2362,63. A su vez, también regula la homeostasis del Ca al modular los canales renales transient receptor potential ion channel (TRPV5)75 y de potasio (K) al regular el canal medular renal outer medullary K (ROMK 176.
- •
Endotelio: el klotho puede inhibir la calcificación vascular, una disminución de klotho se asocia a un aumento en la expresión de los trasportadores de P Pit1/2 y del factor osteogénico Runx2, lo que conllevaría un aumento del trasporte de P en las CMLV y su transformación osteogénica77,78. No obstante, los estudios sobre la expresión de klotho a nivel vascular son contradictorios. Lim et al.79 evidenciaron su expresión en las arterias de individuos sanos; esta expresión se ve descendida en los pacientes con ERC79. Contrariamente, Scialla et al.51 no detectaron expresión de klotho en las CMLV en controles sanos ni en ratones con ERC, tampoco Lindberg et al.80 detectaron niveles de proteína klotho en arterias de ratones wild type. En ratas urémicas, Ritter et al.81 hallaron que la expresión de klotho estaba elevada en la adventicia aórtica y disminuida en la zona íntima media81.
- •
Inhibición de la 1α hidroxilasa que se encarga de la hidroxilación de la 25OH a 1,25(OH)2D317,82: Yoshida et al.82 demostraron en ratones homocigotos para el gen klotho Kl(-/-), que los niveles de 1,25-dihidroxivitamina D estaban aumentados frente a los ratones wild type; a su vez, comprobaron que la expresión del gen 1α hidroxilasa se encontraba aumentado en los ratones Kl (-/-) y que la administración de calcitriol fallaba en inhibir 1α hidroxilasa.
Klotho presenta efectos pleiotrópicos a nivel sistémico: aumenta la transcripción de receptores de eritropoyetina83, aminora el daño provocado por angiotensina II70,84, inhibe la señal insulina/IGF-1, pudiendo provocar resistencia al estrés oxidativo85, posee efectos antifibróticos ya que puede inhibir la señal TGF β86 (factor de crecimiento fibroblástico), y posee efectos antisenescentes y antiapoptóticos87.
Implicaciones de klotho en la ERC y el RCVComo ya se ha mencionado, klotho disminuye precozmente en la ERC19,72. Su déficit podría provocar: calcificación vascular al fomentarse la entrada de P a las CMLV, arterioesclerosis, osteoporosis, calcificación ectópica, envejecimiento prematuro, apoptosis y progresión de la ERC72,78,79,87. Su supresión también implica descenso de la fosfaturia con aumento de P y de los niveles séricos de calcitriol. En el fracaso renal agudo, en modelos experimentales, se ha demostrado el descenso de klotho y, por tanto, su papel como posible biomarcador de fallo renal agudo88,89. Así mismo, su reposición podría conllevar la recuperación del daño renal88,89.
A nivel cardíaco, klotho puede influir directamente sobre la función y el remodelado cardíaco protegiéndolo frente a la HVI. Xie et al.90 valoraron la función cardíaca y la HVI en ratones heterocigotos hipomórficos para el alelo de klotho (Kl/+) con o sin ERC y ratones wild type con o sin ERC. En los ratones con ERC se objetivó un descenso de klotho, HVI y fibrosis miocárdica; todo ello más pronunciado en los Het-klotho. Los ratones sin ERC+Het-klotho también presentaron niveles de klotho menores, pero no datos de HVI. La fracción de eyección estaba reducida significativamente en los ratones ERC+Het-klotho. Al inyectar soluciones de klotho soluble a estos ratones, mejoraban los datos de disfunción miocárdica con independencia de P, FGF23, tensión arterial y FG90.
El mecanismo cardioprotector de klotho se debe a la inhibición del canal TRPC6 (familia de canales de cationes potencial transitoria a subfamilia canónica) que está aumentado en el estado urémico90–92. Ante una agresión cardiológica, TRPC6 permite mayor entrada de Ca al interior celular, se activa la fosfatasa calcineurina, lo que provoca la desfosforilación de NFAT que se transloca al núcleo para inducir la expresión de genes fetales (por ejemplo: β-miosina de cadena pesada), lo cual conlleva la alteración en el remodelamiento cardíaco y la HVI. El gen TRPC6 posee elementos de respuesta frente a NFAT y aumenta su expresión al aumentar el influjo de Ca, causando una activación directa de todo el proceso93,94. Al inhibir el canal TRPC6, klotho podría ser, en un futuro, una medida terapéutica en la HVI.
En la figura 2 esquematizamos la implicación de FGF23, klotho y RCV.
Conclusiones
La búsqueda de marcadores precoces de RCV, inflamación y fibrosis que puedan afectar a la economía corporal y a la función renal es extensa. La EOM-ERC tiene un papel crucial en la salud endotelial y renal; por ello, es prioritario su control y su conocimiento. La investigación en FGF23 y klotho y su relación en ECV abre nuevas expectativas tanto en la prevención como en el tratamiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



