


Cardiovascular factors are one of the main causes of morbidity and mortality in patients with chronic kidney disease. Bone mineral metabolism disorders and inflammation are pathological conditions that involve increased cardiovascular risk in chronic kidney disease. The cardiovascular risk involvement of bone mineral metabolism classical biochemical parameters such as phosphorus, calcium, vitamin D and PTH is well known. The newest markers, FGF23 and klotho, could also be implicated in cardiovascular disease.
Una de las principales causas de morbimortalidad en el paciente con enfermedad renal crónica es la cardiovascular. La inflamación y las alteraciones en el metabolismo óseo mineral son una condición patológica que conlleva aumento del riesgo cardiovascular en la enfermedad renal crónica. Los parámetros bioquímicos clásicos del metabolismo óseo mineral como fósforo, calcio, vitamina D y PTH tienen una implicación muy conocida en el riesgo cardiovascular. Los nuevos marcadores, FGF23 y klotho, también podrían estar implicados en la enfermedad cardiovascular.
Chronic kidney disease (CKD) is a public health problem that is on the rise, with an ever-increasing incidence and prevalence (10% of the general population).1,2 The main cause of mortality in the CKD patient is cardiovascular (CV),3 with an increase in risk of up to 20 times that of the general population, even in initial stages.4–6 Up to 80% of patients with CKD have associated CV disease: hypertension (36%), ischaemic cardiomyopathy (22–39%), atrial fibrillation (30%), valvular heart disease (24%) and left ventricular hypertrophy (LVH) (50–75% in stage 3–4 CKD).7,8
Cardiovascular risk factors in the chronic kidney disease patientThe greater cardiovascular risk (CVR) of patients with CKD is explained by the high presence of classic risk factors and the overlapping of specific factors of a uraemic state as well as by the inflammatory state of CKD; stage 5 adds other factors related to dialysis or transplant that causes an excess of vascular calcification9,10 (Fig. 1). Bone mineral disorder related to CKD (BMD-CKD) plays a crucial role in CKD morbidity and mortality. BMD-CKD includes clinical-chemistry and skeletal abnormalities and extraskeletal calcifications caused by abnormalities in mineral metabolism in CKD. CKD-MBD I presents as one or a combination of the following manifestations11:
- •
Abnormalities in calcium (Ca), phosphate (P), parathyroid hormone (PTH) and vitamin D, klotho, or fibroblast growth factor 23 (FGF23).
- •
Abnormalities in skeletal remodelling, mineralisation, volume, growth or fragility.
- •
Calcifications of cardiovascular or other soft tissues.

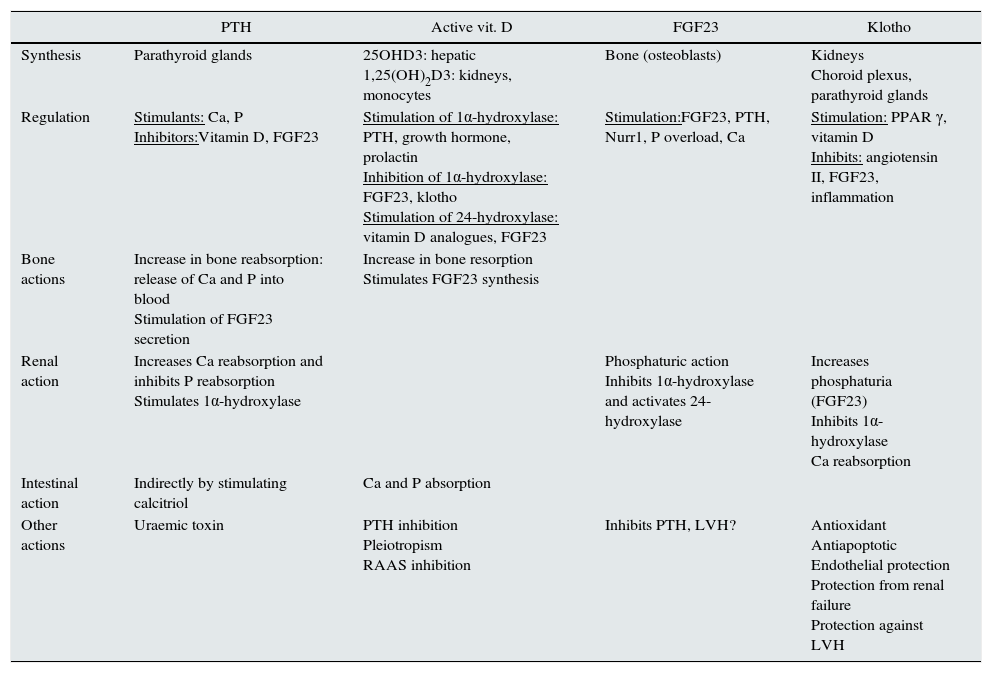
Studying markers of BMD-CKD sometimes allows CVR to be predicted. Table 1 summarises the role of the factors involved in BMD-CKD. The role of klotho and FGF23 in CVD is reviewed below.
Regulation of bone mineral disorder related to chronic kidney disease.
| PTH | Active vit. D | FGF23 | Klotho | |
|---|---|---|---|---|
| Synthesis | Parathyroid glands | 25OHD3: hepatic 1,25(OH)2D3: kidneys, monocytes | Bone (osteoblasts) | Kidneys Choroid plexus, parathyroid glands |
| Regulation | Stimulants: Ca, P Inhibitors:Vitamin D, FGF23 | Stimulation of 1α-hydroxylase: PTH, growth hormone, prolactin Inhibition of 1α-hydroxylase: FGF23, klotho Stimulation of 24-hydroxylase: vitamin D analogues, FGF23 | Stimulation:FGF23, PTH, Nurr1, P overload, Ca | Stimulation: PPAR γ, vitamin D Inhibits: angiotensin II, FGF23, inflammation |
| Bone actions | Increase in bone reabsorption: release of Ca and P into blood Stimulation of FGF23 secretion | Increase in bone resorption Stimulates FGF23 synthesis | ||
| Renal action | Increases Ca reabsorption and inhibits P reabsorption Stimulates 1α-hydroxylase | Phosphaturic action Inhibits 1α-hydroxylase and activates 24-hydroxylase | Increases phosphaturia (FGF23) Inhibits 1α-hydroxylase Ca reabsorption | |
| Intestinal action | Indirectly by stimulating calcitriol | Ca and P absorption | ||
| Other actions | Uraemic toxin | PTH inhibition Pleiotropism RAAS inhibition | Inhibits PTH, LVH? | Antioxidant Antiapoptotic Endothelial protection Protection from renal failure Protection against LVH |
This is a 32-kDa, 251-amino acid protein synthesised and secreted by bone cells, mainly osteocytes. It belongs to the “phosphatonin” hormone group12 owing to its involvement in renal phosphorus elimination. It has been pathophysiologically linked to the so-called “rare hypophosphataemic syndromes”,13 characterised by mineralisation defects and bone deformities, hypophosphataemia, renal P loss and inappropriately low calcitriol levels. It is considered to be one of the main factors in P metabolism regulation.14–16 The biological action of FGF23 depends on the klotho gene14,17–19 which acts as its co-receptor. FGF23 is also expressed in the heart, liver, thyroid gland, parathyroid glands, intestine and skeletal muscle.19
FGF23 regulationFGF23 regulation is determined by:
- 1.
Active vitamin D: calcitriol directly and indirectly increases FGF23 transcription through extracellular signalling pathways mediated by leptin and interleukin 6.20 Calcitriol also increases expression of the nuclear receptor associated with protein 1 (Nurr1) in bone cells and of PTH, which leads to an increase in FGF23.21
- 2.
Ca levels: high calcium stimulate FGF23 secretion.22,23
- 3.
Hyperparathyroidism: the increase in PTH linked to CKD may stimulate FGF23 secretion through Nurr1.19,21 In primary hyperparathyroidism (PHPTH), FGF23 would hypothetically be suppressed since PTH hypersecretion would cause hypophosphataemia so one important stimuli of FGF23 secretion would be reduced.24 However, experimental studies have demonstrated that mice with PHPTH have higher FGF23 levels than controls and that PTH directly stimulate FGF23,25 since FGF23 decreases following parathyroidectomy.25 Yamashita et al.24 demonstrated in patients with PHPTH that FGF23 levels were high compared to healthy controls. However, in this same study, patients without CKD or PHPTH had no differences in FGF23 levels compared to controls,24 and the authors concluded that in PHPTH renal function is a determining factor in FGF23 levels.24
- 4.
P levels: serum P levels have been positively correlated with FGF23 concentration in patients with CKD.26 However, restricting dietary P in patients with CKD has shown contradictory results in FGF23 levels. Some studies have demonstrated that restricting dietary P fails to cause a drop in FGF23 levels in patients with stage 3–4 CKD27 and does not change them in healthy volunteers.28 Despite this, a drop in P absorption with phosphate binders such as sevelamer causes a drop in FGF23 levels.29,30
- 5.
Reduced renal synthesis of klotho: FGF23's affinity for its receptor (especially FGFR1 in the kidneys) is very low.31 Under physiological conditions, when bound to FGFR1, FGF23 would be unable to generate signal transduction.31 In the presence of klotho, FGF23 affinity for its receptor FGFR1 increases significantly and allows its activation with physiological concentration of FGF23.31 A reduction of klotho expression could cause resistance to the action of FGF23; in CKD this resistance would lead to a reduction in the fractional excretion of P and an increase in serum P concentration which in turn stimulate FGF23 secretion.32
Interestingly, iron infusion and low iron levels may induce synthesis of FGF23, although not in its active form.22 Metabolic acidosis, oestrogens and leptin also cause increases in FGF23.33,34
Biological actions of FGF23FGF23 has target receptors called FGFR1, FGFR3 and FGFR4 and the transmembrane receptor β-glucuronidase. To exert its action on FGFR1 in the kidneys it requires its co-receptor klotho.31,35
- •
Bone: in those processes characterised by a mineralisation defect (rickets and osteomalacia) there is excessive production and high activity of FGF23.36 Various groups are researching the direct effect of FGF23 on bone; however, evidence of a direct effect of FGF23 on bone has not yet been demonstrated.37 Various proteins, predominantly in bone, have been found to be involved in regulation of FGF23 synthesis and secretion. These proteins include Phex (P-regulating endopeptidase linked to the X chromosome) and glycoproteins such as that derived from the dentine protein matrix (DMP1) and from the extracellular matrix.36,37 Abnormalities of these proteins cause an increase in FGF23 expression, thereby enhancing phosphaturia resulting in hypophosphataemia and inhibition of bone formation.36,37 Abnormal GLANT expression produces the opposite effect: a decrease in FGF23 activity with an increase in serum P levels and soft tissue calcification (tumoral calcinosis).38
- •
Kidneys: inhibit the expression of type II sodium-phosphate co-transporters (IIa Na/P and IIc Na/P), which causes a decrease tubular reabsorption of P in the proximal tubules thus increasing renal P excretion.17,19,39 It also decreases calcitriol levels by suppressing the activity of the enzyme 1α-hydroxylase (CYP27B1) and stimulating the enzyme 24-hydroxylase (CYP24A1).19,39,40 Finally, in the kidneys, it inhibits transcription of the klotho gene.41
- •
Parathyroid glands: FGF23 decreases PTH production and secretion; this has been demonstrated by several research groups: works such as those by Ben-Dov et al.42 and Krajisnik et al.43 have indicated that FGF23 causes PTH suppression in vivo and in vitro and decreases mRNA expression and transcription and PTH protein secretion.43 However, in hyperplastic uraemic parathyroid glands in rats, FGF23 fails in PTH inhibition compared to healthy parathyroid glands, perhaps owing to a drop in FGFR1 and klotho expression in uraemic hyperplastic glands.44 The impact of parathyroidectomy on FGF23 levels in patients with CKD was studied by Takahashi et al.,45 who designed a study in 30 patients in haemodialysis treated by means of parathyroidectomy with forearm implant and determined FGF23 and klotho levels. They concluded that FGF23 dropped and that klotho had an initial drop and a subsequent increase compared to post-parathyroidectomy values.
- •
Heart: in cardiac muscle cells in mice it has been demonstrated that, through its receptor (FGFR4), FGF23 activates the nuclear factor of activated T cells (NFAT)/calcineurin pathway and causes LVH independently of klotho.46
The patient with end-stage CKD may have FGF23 values of up to 100 times their normal value28; in addition, high FGF23 levels predict CKD progression, as confirmed by several studies .47–49 High FGF23 levels are associated with an increase in mortality adjusted for classic cardiovascular risk factors and other traditional CKD markers.47,50
An association between FGF23 and vascular calcification has been demonstrated, although FGF23 does not seem to induce calcification directly.51 Scialla et al.51 studied the association between FGF23, P, coronary calcification and calcification of the thoracic aorta measured by a CT scan in 1501 participants with CKD (mean glomerular filtration rate [GFR] of 47±17ml/min/1.73m2; stages 2–4). These authors demonstrated that FGF23 was not associated with vascular calcification measured by a CT scan and, in in vitro studies, FGF23 did not cause calcification or induce calcification in of cultured vascular smooth muscle cells (VSMCs), and it is not found in the aorta of mice or humans.51 The role of FGF23 in vascular calcification would be marked by hyperphosphataemia which does induce vascular calcification; VSMCs show osteoblast differentiation in a P-rich media.52,53 It seems to be an association between severity of calcification and FGF23 in CKD, and so FGF23 could be a marker for follow-up and not a factor in the generation of vascular calcification.51
Excess FGF23 leads to an increase in CV morbidity and mortality in CKD54,55 independently of the GFR.56 This could be due:
- 1.
To the fact that FGF23 reduces active vitamin D (calcitriol) levels as it inhibits 1α-hydroxylase and stimulates 24-hydroxylase.19,39
- 2.
To the fact that FGF23 is linked to the presence of markers of inflammation57,58 and oxidative stress such as advanced glycation end products which are associated with vascular calcification.57
- 3.
To the fact that, in some studies, FGF23 has been associated with proteinuria.59
- 4.
To the role of FGF23 in genesis of LVH (a significant source of arrhythmias and heart failure): an elegant study by Faul et al.46 showed, in a cohort of more than 3,000 patients with CKD with a GFR of 20–70ml/min, that there was a correlation between FGF23 levels and LVH. This same study described that FGF23 caused an increase in α-actin in cardiac muscle cells in rats with an increase in expression of LVH markers, such as foetal heavy chain β-myosin, and a drop in adult heavy chain α-myosin. This mechanism is independent of klotho and is mediated by activation of the NFAT/calcineurin pathway.
This is a 130-kDa transmembrane protein that is expressed predominantly in the kidneys (distal, proximal and collecting tubule), parathyroid glands, choroid plexus and endothelium.60,61 It has 3 different forms62,63: klotho-cut, with little biological effect; the complete form, bound to the membrane (which acts as an FGF23 co-receptor); and the secreted form.
Klotho regulation- 1.
In experimental models in vitro it has been demonstrated that the peroxisome proliferator activated receptor γ (PPAR γ) signal64 increases klotho synthesis. Calcitriol also increases expression of klotho in animal models with CKD and high P intake.65
- 2.
Factors that decrease klotho synthesis are: FGF23,66–68 oxidative stress69 and angiotensin II (through its type I receptors and through an increase in tumour necrosis factor-α converting enzyme [TACE]).17,70,71 The klotho gene, which is predominantly synthesised in the kidneys, is decreased in patients with CKD.62,72
- •
Kidneys: klotho directly induces phosphaturia, acting in the proximal tubules by inhibiting type IIa and type IIc Na/P co-transporters.72–74 In addition, klotho is an FGFR-1 co-receptor and, therefore, facilitates the phosphaturic action of FGF23.62,63 At the same time, it also regulates Ca homeostasis by modulating the transient receptor potential ion channel (TRPV5)75 and potassium (K) renal channels by regulating the renal outer medullary K (ROMK1) channel.76
- •
Endothelium: klotho may inhibit vascular calcification; a decrease in klotho is associated with an increase in expression of P transporters PiT1/2 and of the Runx2 osteogenic factor, which would lead to an increase in P transport in VSMCs and their osteogenic transformation.77,78 However, the studies on klotho expression in the vasculature have been contradictory. Lim et al.79 showed its expression in the arteries of healthy individuals; this expression is seen to be decreased in patients with CKD.79 By contrast, Scialla et al.51 did not detect klotho expression in VSMCs in healthy controls or in mice with CKD, and Lindberg et al.80 did not detect klotho protein levels in arteries of wild-type mice. In uraemic rats, Ritter et al.81 found that klotho expression was high in the aortic adventitia and decreased in the intima-media area.81
- •
Inhibition of 1α-hydroxylase which produces hydroxylation of 25-OH to 1,25(OH)2D317,82: Yoshida et al.82 demonstrated in mice homozygous for the Kl(−/−) klotho gene that 1,25-dihydroxyvitamin D levels were increased compared to wild-type mice; at the same time, they confirmed that expression of the 1α-hydroxylase gene was found to be increased in Kl(−/−) mice and that administration of calcitriol failed to inhibit 1α-hydroxylase.
Klotho has pleiotropic effects on a systemic level: it increases transcription of erythropoietin receptors83; reduces the damage caused by angiotensin II,70 inhibits the insulin/IGF-1 signal, which may cause resistance to oxidative stress84; shows antifibrotic effects since it is able to inhibit the transforming growth factor (TGF) β signal85; and has antisenescent and antiapoptotic effects.86
Involvement of klotho in CKD and CVRAs mentioned, klotho decreases early in CKD.19,72 Klotho deficiency may cause: vascular calcification as entry of P into VSMCs is facilitated by klotho deficiency, arteriosclerosis, osteoporosis, ectopic calcification, premature ageing, apoptosis and CKD progression.72,78,79,86 Klotho suppression also leads to a reduction in phosphaturia resulting in hyperphosphatemia and increased serum levels of calcitriol. Studies in experimental models have demonstrated a decrease in klotho in acute renal failure, and therefore klotho levels may be a potential biomarker of acute renal failure.87,88 Likewise, some authors propose that replacement of klotho may help a recovery from acute renal damage.87,88
In the heart, klotho may directly influence cardiac function and remodelling by protecting it against LVH. Xie et al.89 assessed cardiac function and LVH in heterozygous mice hypomorphic for the (Kl/+) klotho allele with or without CKD and wild-type mice with or without CKD. In mice with CKD, a drop in klotho, LVH and myocardial fibrosis was observed; all this was less pronounced in klotho-heterozygous mice. Klotho-heterozygous mice without CKD also had lower klotho levels, but no evidence of LVH. Ejection fraction was significantly reduced in klotho-heterozygous mice with CKD. When soluble klotho was injected into these mice, myocardial dysfunction improved independently of P, FGF23, blood pressure and GFR.89
The cardioprotective effect of klotho may be due to the inhibition of TRPC6 channel (transient receptor potential family of cation channels in canonical subfamily) which is increased in a uraemic state.89–91 When facing cardiological aggression, TRPC6 allows greater entry of Ca into the cell, which produces activation of the phosphatase calcineurin. This causes dephosphorylation of NFAT that translocates to the nucleus to induce expression of foetal gene (for example: heavy chain β-myosin). This leads to abnormal cardiac remodelling and LVH. The TRPC6 gene posses response elements against NFAT and its expression is increased with cellular influx of Ca, causing direct activation of the entire process.92,93 In the future, if the TRPC6 channel is inhibited, klotho could be used as a potential therapeutic strategy against LVH.
Fig. 2 outlines the involvement of FGF23 and klotho on CVR.
Conclusions
The search for early markers of CVR, inflammation and fibrosis that may affect body economy and renal function is extensive. BMD-CKD plays a crucial role in endothelial and renal health, and so knowledge and management of it is a priority. Research on FGF23 and klotho and their relationship to CVD is opening new expectations in both prevention and treatment.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Salanova Villanueva L, Sánchez González C, Sánchez Tomero JA, Aguilera A, Ortega Junco E. Enfermedad óseo mineral relacionada con la enfermedad renal crónica: Klotho y FGF23; implicaciones cardiovasculares. Nefrología. 2016;36:368–375.



