








El sistema del Complemento protege al organismo frente a procesos infecciosos, tumorales y autoinmunes, y requiere una regulación muy estricta para evitar una activación excesiva o inespecífica. Entre los componentes reguladores del Complemento destaca el factor H (FH), que controla su activación en plasma y sobre la superficie de las células y tejidos propios. FH está relacionado evolutiva y estructuralmente con un conjunto de proteínas plasmáticas denominadas FHRs (FH-Related proteins), que podrían actuar como antagonistas funcionales de FH. Numerosos estudios realizados en pacientes de Síndrome Hemolítico-Urémico atípico (SHUa), glomerulopatía C3 (GC3), y nefropatía por IgA (NIgA) han identificado variantes genéticas raras que alteran sustancialmente la función del FH y las proteínas FHRs, y contribuyen de forma muy relevante a la predisposición genética a estas patologías. Estos pacientes presentan también una mayor frecuencia de determinados polimorfismos cuya repercusión en el mecanismo patogénico se está empezando a dilucidar. En los últimos años, la disponibilidad de reactivos específicos para cuantificar las proteínas FHRs de forma fiable en controles y pacientes, ha mostrado que algunos de los polimorfismos asociados a SHUa, GC3 o NIgA determinan cambios en los niveles plasmáticos de FH y proteínas FHRs, que podrían repercutir en la correcta regulación de la activación del Complemento y contribuir así al desarrollo de estas patologías.
The complement system is a first line of defence against infectious, tumoral or autoimmune processes, and it is constitutively regulated to avoid excessive or unspecific activation. Factor H (FH), a most relevant complement regulator, controls complement activation in plasma and on the cellular surfaces of autologous tissues. FH shares evolutionary origin and structural features with a group of plasma proteins known as FH-Related Proteins (FHRs), which could act as FH functional antagonists. Studies in patient cohorts of atypical Haemolytic-Uraemic Syndrome (aHUS), C3 Glomerulopathy (C3G), and IgA nephropathy (IgAN), have identified rare genetic variants that give rise to severe FH and FHRs dysfunctions, and are major genetic predisposing factors. These patients also have a higher frequency of a few polymorphisms whose relevance as disease risk factors is incompletely understood. In the last years, the availability of specific reagents has allowed a more precise quantitation of FH and FHRs in plasma samples from patients and controls. These studies have revealed that some aHUS, C3G or IgAN risk polymorphisms determine mild changes in FH or FHRs levels that could somehow perturb complement regulation and favour disease pathogenesis.
El Sistema del Complemento es esencial para la respuesta inmune innata, y modula también la inmunidad adaptativa porque favorece la generación de anticuerpos y la memoria inmunológica. Además de proteger al organismo de forma inmediata frente a una gran variedad de patógenos, el Complemento juega un papel clave en la eliminación de los inmunocomplejos y células propias dañadas, que podrían provocar daño tisular y favorecer procesos autoinmunes1.
El Complemento lo integran más de 30 proteínas plasmáticas o de membrana sintetizadas constitutivamente, que ejercen una función efectora o una función reguladora intrínsecas a la homeostasis del sistema. Todo el funcionamiento del Complemento pivota alrededor del proceso de activación del componente C3, una de las proteínas plasmáticas más abundantes (fig. 1). La activación del C3 es una reacción proteolítica por la que la molécula de C3 se escinde en los fragmentos C3b y C3a, y a la que se puede llegar a través de 3 vías en las que participan distintos componentes: la Vía Clásica (VC), la Vía Alternativa (VA), y la Vía de las Lectinas (VL). La generación de C3b, a su vez, conduce a la etapa final de activación del Complemento, conocida como Vía Lítica2.
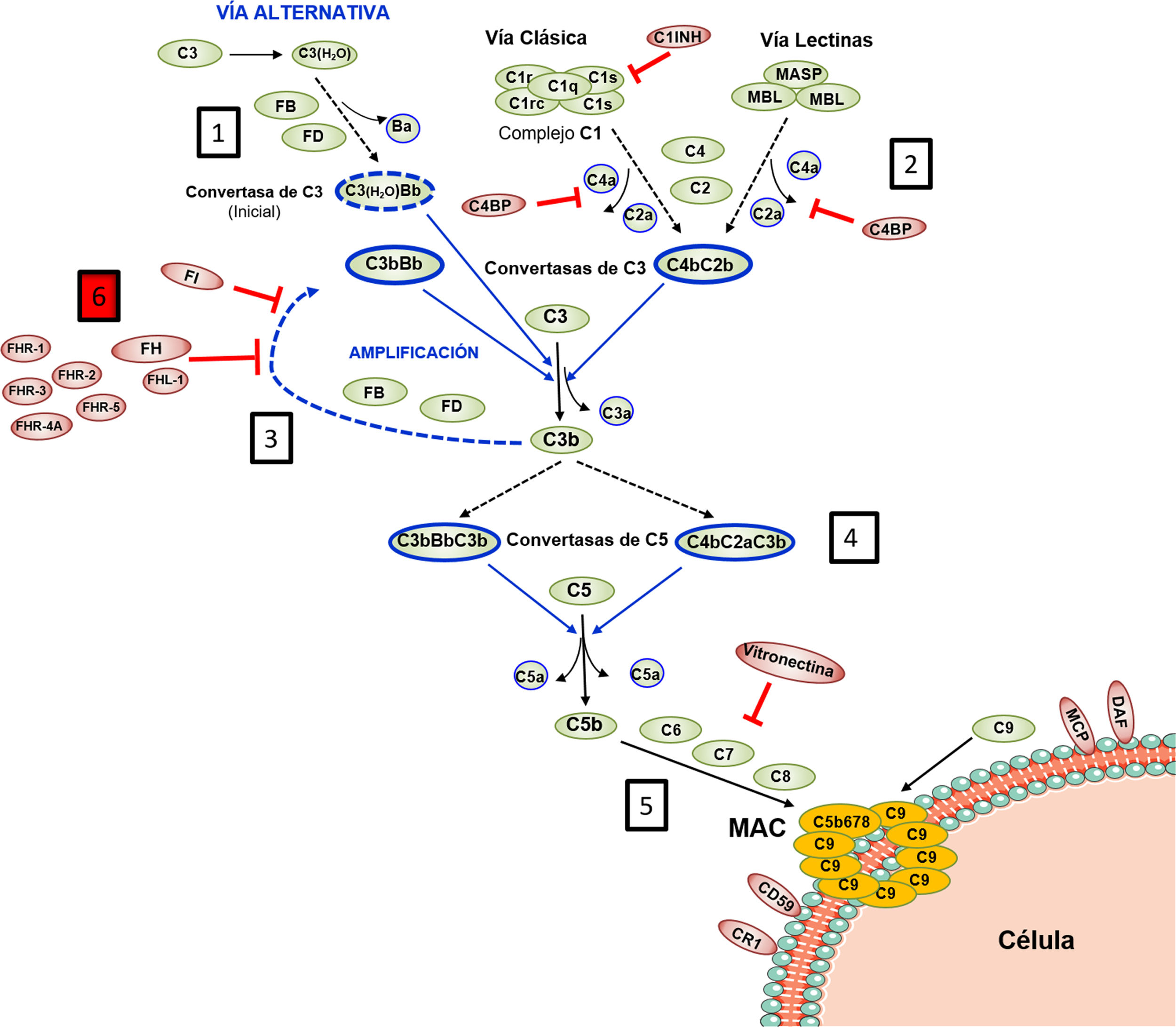
Activación y Regulación del Complemento
El Complemento es un sistema autorregulado cuya piedra angular es la activación proteolítica del componente C3. 1) La hidrólisis espontánea de algunas moléculas de C3 conduce a la activación basal de la Vía Alternativa (VA). 2) El Complemento puede activarse de forma más intensa a través de la Vía Clásica (VC) y de la Vía de las Lectinas (VL), cuando los componentes C1q o MBL/Ficolinas reconocen complejos antígeno-anticuerpo solubles, o moléculas de carbohidratos presentes en determinadas superficies (células propias dañadas o microorganismos). Esta activación conduce a la formación de una Convertasa de C3 (C4bC2b), que escinde el C3 en los fragmentos C3b y C3a. 3) La generación de C3b da lugar a otra Convertasa de C3 (C3bBb) y a la amplificación de la activación basal de la VA, que se convierte entonces en el mecanismo más eficaz de activación del Complemento. 4) El siguiente paso es la activación del componente C5, generando los fragmentos C5b y C5a. 5) La última etapa es la denominada Vía Lítica, que da lugar a la formación de un complejo multimolecular (MAC, Membrane Attack Complex) que se inserta en la membrana de la superficie activadora y forma poros que la lisan por desequilibrio osmótico. 6) El proceso de activación del Complemento está regulado en todas sus etapas por múltiples proteínas (representadas en color rojo) que evitan que se active sobre nuestras propias células y tejidos; la regulación de la VA depende en gran medida del regulador Factor H y de su isoforma FHL-1. El rol preciso de las proteínas FHR (homólogas estructurales de FH) en el proceso de activación-regulación del Complemento no se conoce en detalle. Para la nomenclatura del Complemento ver las referencias49,50.
Las características bioquímicas del proceso de activación del Complemento lo convierten en una herramienta sumamente rápida y eficaz en la defensa frente a patógenos, pero también pueden resultar perjudiciales para el huésped. Así, una activación excesiva puede llevar a un rápido consumo de componentes que facilita una nueva invasión por patógenos, y provocar una respuesta inflamatoria exacerbada. Por otro lado, los productos de activación del Complemento pueden depositarse sobre las células propias y dañarlas. En situaciones fisiológicas, estas consecuencias adversas se evitan gracias a que todas y cada una de las distintas etapas del proceso de activación del Complemento están controladas por componentes reguladores, algunos presentes en plasma y otros localizados en la superficie de la mayoría de las células y tejidos propios. En consecuencia, para que el Complemento sea eficaz y específico, y no genere daño autólogo, el equilibrio entre su activación y regulación es fundamental3.
La regulación de la VA del Complemento es especialmente relevante. Esta vía de activación, evolutivamente la más antigua, está permanentemente activada en el plasma debido a la hidrólisis espontánea de un enlace tioéster interno en algunas moléculas de C3. Normalmente, esta activación basal de la VA no supera un umbral perjudicial, gracias a la existencia de componentes del Complemento que la regulan. Por este motivo, cualquier situación, genética o adquirida, que altera la regulación de la VA puede provocar daño autólogo por el Complemento, siendo el riñón uno de los órganos más vulnerables4. Las patologías renales comúnmente asociadas a la desregulación de la VA del Complemento son el Síndrome Hemolítico-Urémico atípico (SHUa), la glomerulopatía C3 (GC3) y la nefropatía por IgA (NIgA). Muchos de estos pacientes tienen variantes genéticas patogénicas o autoanticuerpos circulantes que afectan al normal funcionamiento del Factor H (FH), principal regulador de la VA del Complemento, y que es capaz de controlar la activación de C3 tanto en plasma como sobre las superficies celulares propias; no menos relevante es la existencia de variantes genéticas comunes (i.e, polimorfismos) en el gen del FH que también contribuyen al desarrollo y/o expresividad clínica de estas patologías5. En la primera parte de esta revisión recapitularemos las variantes patogénicas y los polimorfismos que afectan a la regulación de la VA del Complemento dependiente de FH, para posteriormente centrarnos en la repercusión que los niveles de FH y de sus proteínas homólogas, Factor H-Related Proteins (FHRs), pueden tener en patología renal.
Familia de proteínas FH y FHRsFH es una glicoproteína plasmática de 150kDa, formada por 20 dominios estructurales denominados SCRs (Short Consensus Repeats) o CCPs (Complement Control Proteins). En la molécula de FH se distinguen dos regiones funcionalmente muy relevantes: la región N-terminal (SCRs 1-4), donde reside su capacidad para regular la activación del Complemento en plasma, y la región C-terminal (SCRs 19-20), que permite a FH unirse transitoriamente a las superficies celulares propias y protegerlas del daño indiscriminado por el Complemento6.
FH está codificado por el gen CFH, que genera también una variante de splicing alternativo denominada Factor H-like protein 1 (FHL-1). La proteína FHL-1 contiene únicamente los dominios SCRs 1-7 de FH, seguidos de una cola de 4 aminoácidos; como consecuencia, FHL-1 puede regular el Complemento como FH, pero no puede unirse a las superficies celulares propias7. Junto al gen CFH, en el agrupamiento génico Regulators of Complement Activation (RCA) del cromosoma 1, se encuentran los 5 genes CFHRs, que codifican las 5 proteínas homólogas a FH denominadas colectivamente FHRs8. Los genes CFH-CFHRs tienen regiones exónicas e intrónicas muy similares, que reflejan una historia evolutiva compartida, y caracterizada por una sucesión de eventos de duplicación génica (fig. 2a). Este origen común explica la elevada similitud estructural de FH y las proteínas FHRs, que también están formadas enteramente por dominios SCRs, algunos de los cuales alcanzan el 100% de identidad de aminoácidos con los dominios homólogos en FH. El distinto tamaño molecular de FH y las proteínas FHRs permite que todas ellas se puedan identificar fácilmente mediante un análisis de Western-blot en una muestra de plasma o suero del paciente (fig. 2b).
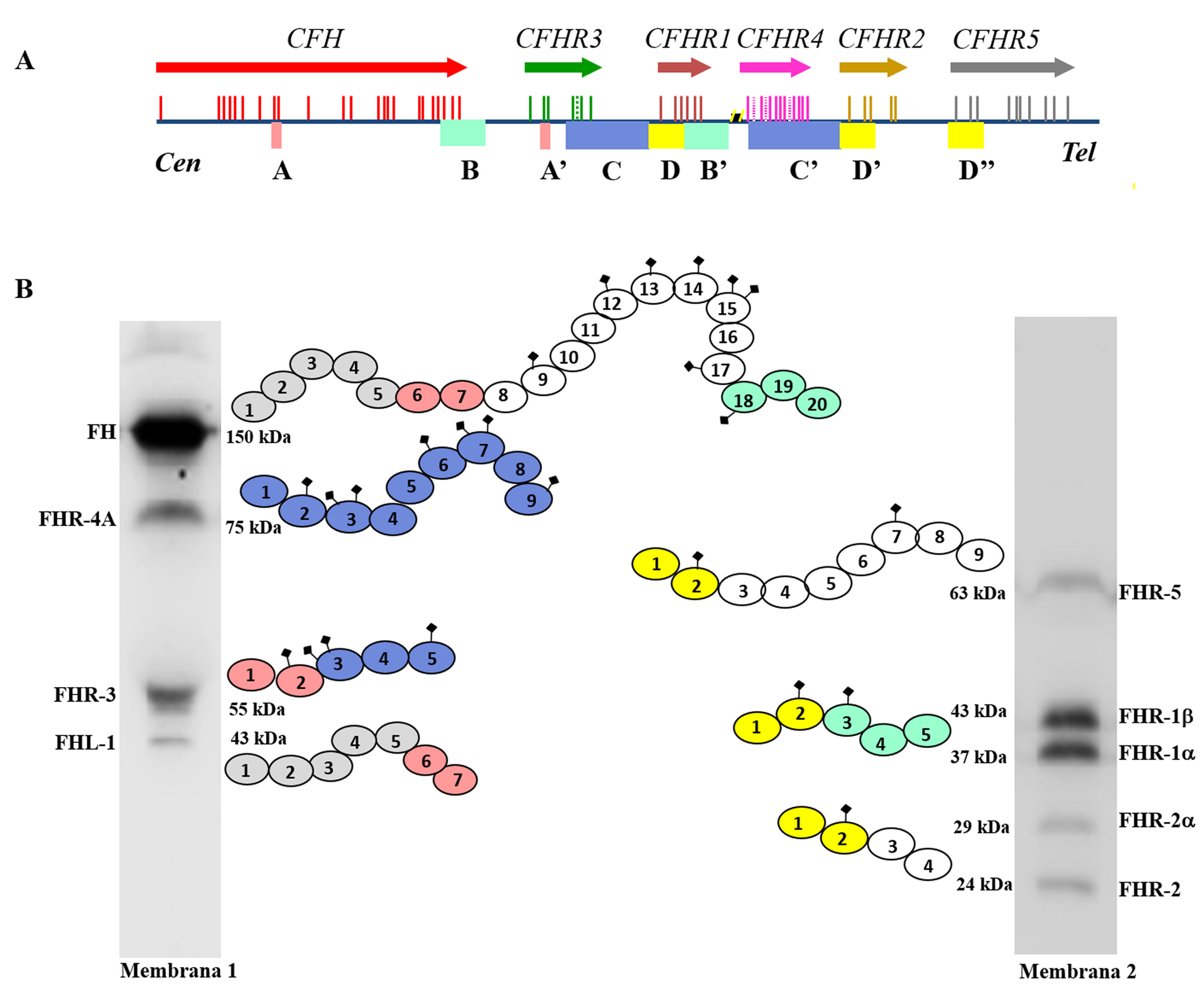
Familia de factor H y proteínas FHRs
A) Región del agrupamiento génico RCA (Regulator of Complement Activation, 1q32-33) que contiene el gen CFH y los 5 genes CFHRs. Todos los genes tienen la misma orientación 5’-3’. Las letras A, B, C y D denotan grupos exón-intrón homólogos entre sí. B) Identificación de FH, FHL-1 y las 5 proteínas FHRs de un suero humano normal mediante análisis de Western-blot con 2 preparaciones de anticuerpos con distinta especificidad. Todas las proteínas están formadas por 4-20 dominios globulares homólogos; los dominios con el mismo color son los más similares entre sí, y están codificados por las duplicaciones A, B, C o D de los genes respectivos.
Las proteínas FHRs pueden interaccionar con C3b y unirse a ligandos de FH localizados en la superficie celular. Sin embargo, ninguna proteína FHR tiene dominios SCRs homólogos a la región N-terminal del FH, por lo que no pueden regular la activación del Complemento en plasma de la misma manera, ni proteger a las superficies celulares propias frente al Complemento autólogo. Estas características llevaron a proponer que las proteínas FHRs actuaban como desreguladores del FH9, pero su función precisa no está bien caracterizada y es objeto de controversia10. En este sentido, no se puede descartar que algunas proteínas FHRs tengan cierta actividad reguladora del Complemento11,12, pero también se ha demostrado que pueden actuar como activadores del mismo. Es el caso de las proteínas FHR-1, FHR-4 y FHR-5, que pueden unirse a C3b, promover la formación de la Convertasa de C3 y activar la VA, y también pueden activar la VC a través de su unión a las pentraxinas PTX3 y Proteína C reactiva13–17.
Puesto que las proteínas FHRs desregulan el FH y/o potencian directamente la activación de la VA del Complemento, su función fisiológica es antagónica a la del FH. Por tanto, las variantes genéticas que alteran la función de FH y las proteínas FHRs, o que producen cambios en sus niveles plasmáticos, pueden desregular la VA y predisponer al desarrollo de patología renal.
Variantes genéticas CFH/CFHRs en SHUa, GC3 y NIgAComo se ha mencionado, el equilibrio funcional entre el FH y las proteínas FHRs contribuye a mantener la homeostasis de la VA del Complemento y evitar el desarrollo de patologías renales como el SHUa, la GC3 o la NIgA. Los estudios realizados en distintas cohortes de pacientes de SHUa y GC3 han identificado variantes patogénicas de FH y proteínas FHRs que perturban la regulación de la VA, y que se detallan en revisiones recientes18,19. También se han descrito algunas variantes patogénicas en pacientes de NIgA20,21.
Muchas de estas variantes patogénicas resultan de la existencia de duplicaciones exón-intrón en la región CFH-CFHRs, que la hacen muy propensa a eventos de conversión génica y recombinación homóloga desigual que originan mutaciones, duplicaciones, deleciones o genes híbridos. La mayoría de estos reordenamientos anómalos no pueden detectarse mediante secuenciación estándar del ADN, por lo que se precisan técnicas capaces de determinar el número de copias de la región que se sospecha pueda estar duplicada o ausente, o incluso técnicas mucho más específicas que sólo se realizan en algunos laboratorios de investigación y requieren mucho tiempo. Algunos de estos reordenamientos anómalos provocan la deficiencia total de una o varias proteínas, mientras que otros generan proteínas que tienen un tamaño molecular diferente de las proteínas nativas; estas 2 situaciones se detectan rápidamente mediante un sencillo análisis de Western-blot(fig. 3) que, además, proporciona una información valiosa para diseñar estrategias genéticas específicas para caracterizar el defecto genético subyacente.
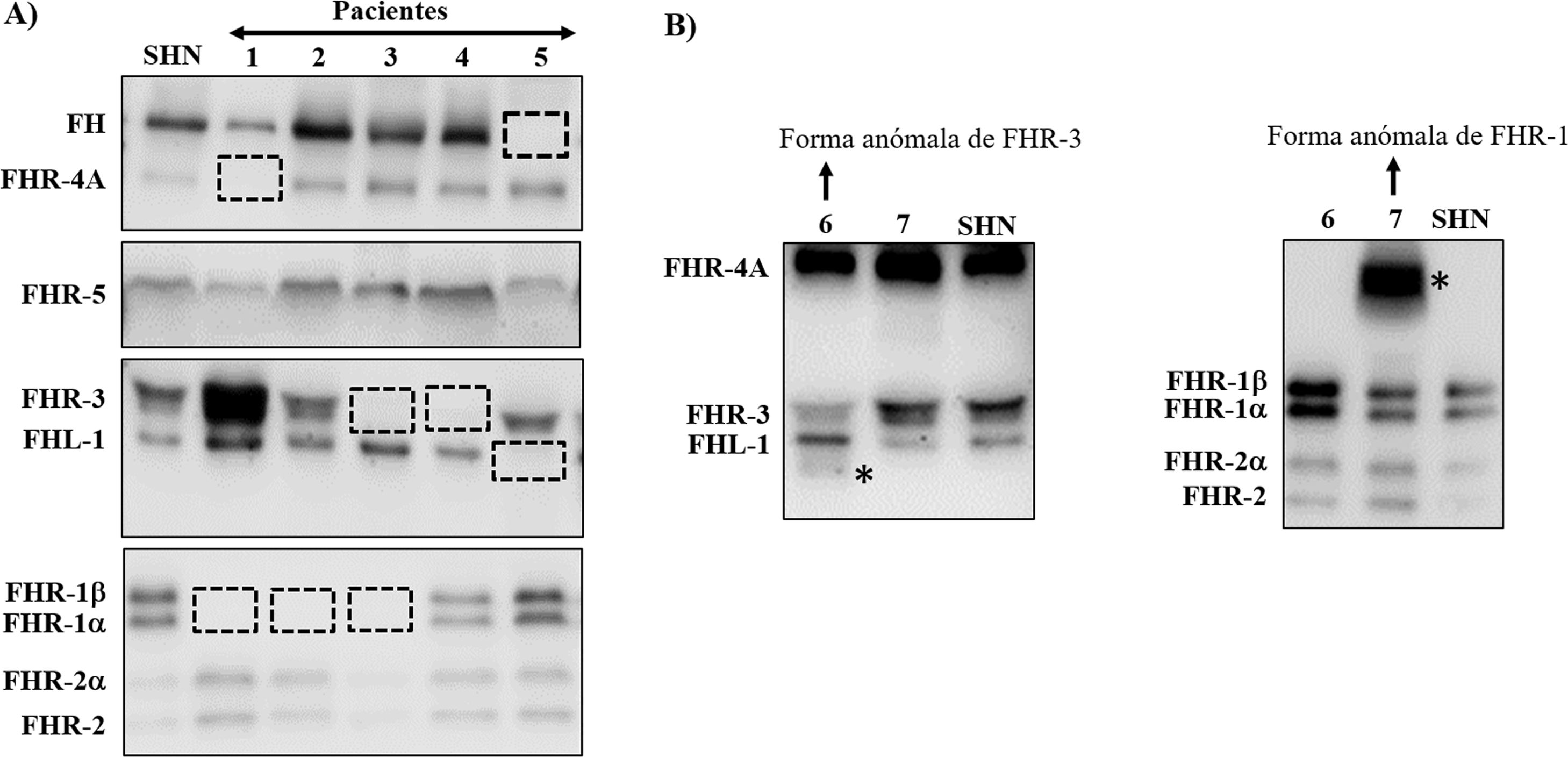
Screening de deficiencias y formas anómalas de FH y proteínas FHRs en pacientes de SHUa y GC3 mediante Western-blot
A) Patrones de FH y FHRs en un individuo sano (SHN) y en 5 pacientes de SHUa y GC3 que tienen deficiencia completa de 1 o 2 proteínas; los cuadrados en línea discontinua señalan la posición de la proteína que falta. Las diferencias en la intensidad de otras bandas reflejan que existe una importante variabilidad interindividual en los niveles de FH y proteínas FHRs, cuya relevancia fisiopatológica no está suficientemente clara.
B) Patrones de FH y FHRs en 2 pacientes que presentan bandas anómalas (señaladas con un asterisco), cuyo tamaño no se corresponde con el de ninguna proteína nativa. Análisis de WB adicionales y estudios genéticos identificaron que la banda anómala del paciente 6 corresponde a una isoforma corta de FHR-3, y la del paciente 7 a una forma de FHR-1 parcialmente duplicada.
Además de variantes patogénicas raras, la región CFH-CFHRs contiene multitud de polimorfismos (i.e. variantes genéticas con una frecuencia en la población mayor del 1%), algunos de los cuales se asocian a patología renal (tabla 1). Entre estos polimorfismos destaca la deleción de los genes CFHR3 y CFHR1 (ΔCFHR3-CFHR1), que cuando se encuentra en homocigosis se asocia con riesgo a SHUa22 y protección frente a NIgA23. Un 80% de los pacientes de SHUa que desarrollan autoanticuerpos anti-FH son homocigotos para la variante ΔCFHR3-CFHR1 y, por consiguiente, carecen de las proteínas FHR-3 y FHR-1.
Variantes genéticas comunes en la región CFH-CFHRs y patología renal
| Variante | Consecuencia funcional | Asociación | Referencia |
|---|---|---|---|
| CFH(H1) | Contiene la variante His402, que disminuye la unión de FH a diferentes carbohidratos de membrana | Riesgo GC3 | 24 |
| CFH(H2) | Contiene la variante Ile62 que incrementa la actividad reguladora de FH. | Protección SHUa y GC3 | 24 |
| CFH(H3) | Se asocia con niveles más bajos de FH | Riesgo SHUa | 25 |
| CFH(H4) | Se asocia con la variante ΔCFHR3-CFHR1Niveles de FH más altos | Riesgo SHUa(HOM) | 46 |
| ΔCFHR3-CFHR1 | Se asocia con autoanticuerpos anti-FH | Riesgo SHUa (HOM) | 26 |
| La deficiencia de FHR-1 favorece la regulación de FH | Protección IgAN | 23 | |
| CFHR3*B | Se asocia con niveles más elevados de FHR-3 | Riesgo SHUa | 27 |
| CFHR1*B | Contiene la secuencia Tyr157-Val159-Gln175, que podría aumentar la competencia de FHR-1 con FH. | Riesgo SHUa | 26 |
| CFHR4*B | Contiene la variante Pro405, pero se desconoce su repercusión funcional. | Riesgo SHUa | 28 |
| CFHR5Ser62 | Afecta a la interacción de FHR-5 con C3b y podría influir en la activación del complemento | Riesgo GC3 | 29 |
El gen CFH contiene varios polimorfismos de un solo nucleótido (Single Nucleotide Polymorphisms, SNPs) que se encuentran en desequilibrio de ligamiento y definen diferentes haplotipos, siendo los más frecuentes CFH(H1), CFH(H2), CFH(H3) y CFH(H4). El haplotipo CFH(H1), que contiene la variante FHHis402, se asocia con riesgo a GC3, mientras que el haplotipo CFH(H2), que incluye la variante FHVal62, se considera un factor de protección frente a SHUa y GC324. El haplotipo CFH(H3) se asocia con riesgo a SHUa y con menores niveles de FH25. Finalmente, el haplotipo CFH(H4) está en desequilibrio de ligamiento con la deleción ΔCFHR3-CFHR1, y es más frecuente en los pacientes de SHUa con autoanticuerpos anti-FH.
Los genes CFHR1 y CFHR3 también tienen varios SNPs en desequilibrio de ligamiento que configuran 2 alelos mayoritarios: CFHR1*A/CFHR1*B26 y CFHR3*A/CFHR3*B25. Los alelos CFHR1*B y CFHR3*B se asocian con riesgo a SHUa. El alelo CFHR3*B determina mayores niveles de la proteína FHR-327, mientras que el alelo CFHR1*B no parece tener relación con los niveles de la proteína FHR-1; en este caso, su asociación con SHUa se debe probablemente a que presenta una mayor identidad de aminoácidos con FH y por tanto podría competir más eficazmente por la unión a los mismos ligandos.
Es interesante que el haplotipo de riesgo a SHUa CFH(H3) se presente mayoritariamente en el mismo cromosoma que los alelos CFHR3*B y CFHR1*B, dando lugar a un haplotipo extendido CFH(H3)-CFHR3*B-CFHR1*B que probablemente determina un aumento en los niveles relativos FHR-3/FH que podría favorecer el desarrollo del SHUa. El haplotipo CFH(H1), por su parte, se presenta habitualmente con los alelos CFHR3*A y CFHR1*A, formando el haplotipo extendido CFH(H1)-CFHR3*A-CFHR1*A, que es mayoritario en la población control sana.
En el gen CFHR4 también se ha descrito un polimorfismo aparentemente asociado con SHUa, y que no parece estar relacionado con los niveles de la proteína FHR-4A28, y el gen CFHR5 contiene varios polimorfismos que se asocian con mayor riesgo a GC329.
En los pacientes de NIgA, la región CFH-CFHRs no se ha caracterizado con el mismo detalle que en los pacientes de SHUa y GC3. Por este motivo, no puede descartarse que haya otros polimorfismos asociados a NIgA, aparte de la ya mencionada deleción ΔCFHR3-CFHR1.
Niveles de FH y proteínas FHRs en controles y pacientesLa actividad de la VA del Complemento también puede verse afectada por cambios en los niveles fisiológicos de FH y proteínas FHRs, como demuestran los trabajos que comentaremos en este apartado, en el que analizaremos además los polimorfismos en la región CFH-CFHRs que afectan a los niveles de proteína. Es importante señalar que la mayoría de estos estudios expresan los niveles de proteína en μg/ml; puesto que el tamaño molecular de FH (150 kDa) es mayor que el de cualquiera de las proteínas FHRs (entre 24kDa y 75 kDa), para poder interpretar correctamente los cambios en los niveles plasmáticos de FH y proteínas FHRs es preciso comparar las concentraciones de todas ellas en base molar (fig. 4).
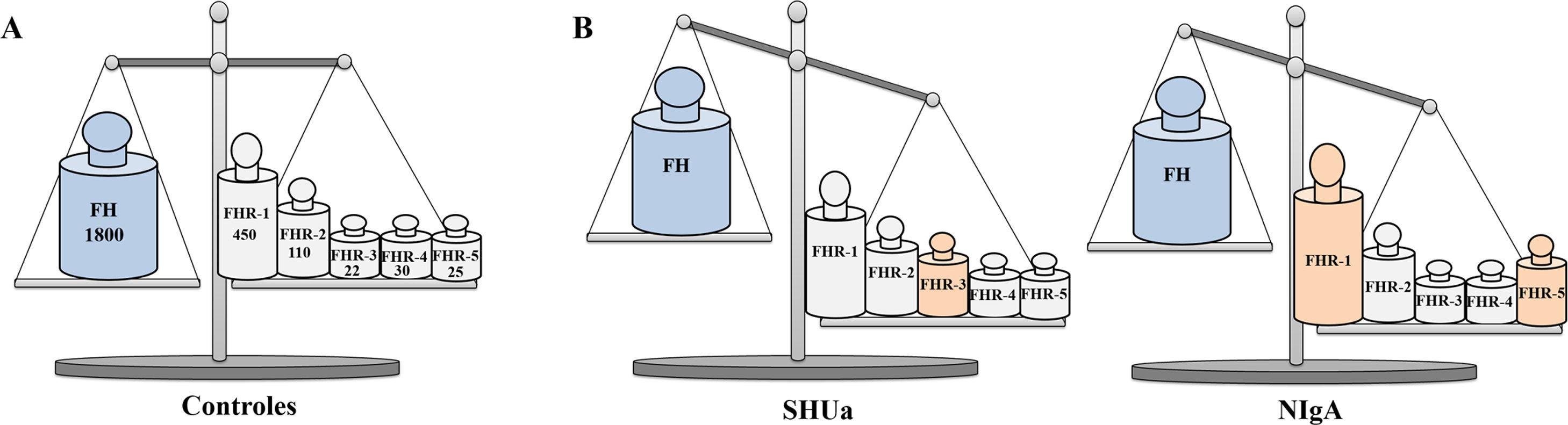
Relevancia fisiopatológica de los niveles plasmáticos de FH y proteínas FHRs
Representación gráfica de los niveles de FH y proteínas FHRs en Controles, pacientes de SHUa y pacientes de NIgA. A) En controles sanos, los niveles de FH y los de las proteínas FHRs están en equilibrio funcional. Los números debajo del nombre de cada proteína expresan su concentración media en base nanomolar33,40,43, por lo que equivalen al número de moléculas que hay de cada una de ellas en un determinado volumen de plasma. B) Pequeños aumentos en la concentración de alguna proteína FHR pueden desequilibrar la regulación de la VA del Complemento por el FH, y favorecer el desarrollo de patologías renales; en algunos pacientes de SHUa aumentan los niveles de FHR-3,27 mientras que en algunos pacientes de NIgA aumentan los niveles de FHR-1 y FHR-5.21,34.
FH es una proteína relativamente abundante en plasma, con un rango de concentración amplio (116-562μg/ml) que está determinado genéticamente en un 62%, aumenta ligeramente con la edad, y disminuye por el consumo de tabaco30. Los valores medios en controles adultos son aproximadamente 250μg/ml 21,31–35, similares a los observados en controles pediátricos36 y mayores que en neonatos37. Los niveles de FHL-1 (la isoforma corta de FH), por su parte, son entre 3 y 40 veces menores que los de FH, en base molar7,38.
La elevada similitud entre FH y las proteínas FHRs ha supuesto un hándicap a la hora de generar anticuerpos específicos para cuantificar cada una de estas proteínas mediante ensayos ELISA Por este motivo, se han utilizado otras técnicas (Western-blot semicuantitativo, espectrometría de masas) que han proporcionado resultados muy dispares10. En los últimos años, varios grupos de investigación han generado anticuerpos específicos para todas las proteínas FHRs, lo que está permitiendo determinar su concentración plasmática de forma más fiable. Actualmente existen anticuerpos comerciales para todas las proteínas FHRs, excepto la proteína FHR-3, de manera que es posible diseñar ensayos ELISA específicos para cuantificarlas. No obstante, el hecho de que no haya un protocolo común, y la falta de reactivos de referencia hace que los niveles obtenidos en estos ensayos sean a veces muy diferentes. Una complicación añadida es la capacidad de FHR-1, FHR-2 y FHR-5 para formar complejos diméricos. Esta peculiaridad se debe a que los dominios SCR1-SCR2 de estas 3 proteínas son muy similares entre sí y contienen aminoácidos que les permiten forman homodímeros y heterodímeros que tienen mayor capacidad para competir con FH39; la cuantificación de todas estas formas se ha abordado en un estudio realizado en controles holandeses, según el cual no habría formas monómericas de estas proteínas circulando en plasma40.
Los niveles de las proteínas FHR-1 y FHR-3 están determinados, en primer lugar, por la existencia de la variante genética ΔCFHR3-CFHR1, ya que los individuos homocigotos para esta variante no tienen ninguna copia de los genes CFHR1 y CFHR3, y los individuos heterocigotos sólo tienen una. Como la frecuencia de la variante genética ΔCFHR3-CFHR1 varía mucho entre poblaciones, se trata de un rasgo genético muy relevante cuando se quieren comparar los niveles de FHR-1 y FHR-3 entre distintas cohortes de controles sanos, o entre controles y pacientes de etnias diferentes. Así, un 5% de los individuos europeos caucasoides no tiene ninguna copia de los genes CFHR1 y CFHR3, pero esta frecuencia se eleva al 33% en individuos de Nigeria, mientras que en japoneses y sudamericanos es prácticamente cero41. En este apartado nos referiremos a los datos obtenidos en poblaciones caucasoides.
Los niveles de FHR-1 en individuos con una sola copia del gen CFHR1 (i.e. heterocigotos para la variante ΔCFHR3-CFHR1) son aproximadamente la mitad que en los que tienen dos copias del mismo (61μg/ml vs 122μg/ml)21. Estos resultados coinciden bastante bien con los niveles de 95μg/ml observados en otra población control europea, en la que no se tuvo en cuenta el número de copias de la variante ΔCFHR3-CFHR134. Sin embargo, el estudio en controles holandeses, que además discrimina entre homodímeros FHR-1/1 y heterodímeros FHR-1/2, obtiene unos valores totales de FHR-1 unas 10 veces menores40. Los niveles plasmáticos de la proteína FHR-3 son bastante inferiores a los de FH y FHR-1, pero, como cabría esperar, también son aproximadamente el doble en los controles adultos que tienen 2 copias del gen CFHR3 que en los que sólo tienen una (0.83μg/ml vs 0.38μg/ml)33. En la cuantificación de FHR-3, no obstante, hay que tener en cuenta un componente genético adicional, ya que el alelo CFHR3*B da lugar a mayores niveles de proteína que el alelo CFHR3*A27; por tanto, los niveles de FHR-3 en plasma van a depender en última instancia del número de copias de los alelos CFHR3*A y CFHR3*B que tenga cada individuo, lo que explica la gran variabilidad de niveles de FHR-3 (entre 0.14 y 1.16μg/ml) observada en una pequeña cohorte control española en la que se caracterizaron las variantes ΔCFHR3-CFHR1, CFHR3*A y CFHR3*B27. En controles pediátricos, los niveles de FHR-1 son ligeramente inferiores a los de adultos, mientras que en los de FHR-3 no se observan diferencias36.
La concentración media de la proteína FHR-2 en el plasma de controles adultos es de unos 6μg/ml, que corresponden mayoritariamente a heterodímeros FHR-1/2 (5.2μg/ml) y en mucha menor proporción (0.8μg/ml) a homodímeros FHR-2/240. Los niveles de FHR-2 en controles pediátricos son muy similares a los observados en adultos36. No se han descrito polimorfismos del gen CFHR2 que afecten a los niveles de expresión de la proteína.
La proteína FHR-4A es la isoforma mayoritaria del gen CFHR4, que origina también un producto de splicing alternativo denominado FHR-4B, de menor tamaño que la isoforma FHR-4A42. Las primeras cuantificaciones no distinguían las isoformas FHR-4A y FHR-4B, y estimaban unos niveles medios de proteína de 25.4μg/ml14. El desarrollo de anticuerpos específicos ha permitido cuantificar las 2 isoformas de forma individual43; los resultados muestran que los niveles de la isoforma FHR-4A son 2.55 ug/ml (10 veces menores de los estimados anteriormente), mientras que la isoforma FHR-4B no se detecta en plasma, lo que suscita dudas razonables sobre si realmente se secreta. Los niveles de FHR-4A no parecen estar asociados con la edad, pero sí son ligeramente menores en controles pediátricos que en adultos36. Recientemente se ha descrito un polimorfismo en el intrón 1 del gen CFHR4 que se asocia con niveles más elevados de la isoforma FHR-4A35.
La proteína FHR-5, al igual que FHR-1 y FHR-2, también tiene 2 dominios de dimerización, pero aparentemente sólo circula en plasma en forma de homodímeros FHR-5/5. Los niveles medios de FHR-5 en controles adultos holandeses son 1.66μg/ml40, algo inferiores a los reportados anteriormente en otras cohortes, que oscilan entre 2.5 y 5.5μg/ml34,44,45. Los niveles de FHR-5 son más bajos en niños menores de 3 años, edad a la que alcanzan prácticamente los mismos valores que en adultos36.
¿Cómo varían los niveles de FH y proteínas FHRs en pacientes de SHUa, NIgA y GC3? Como mencionamos anteriormente, algunos polimorfismos que inciden directamente en los niveles de proteína tienen frecuencias diferentes en pacientes y en controles. Es el caso del haplotipo CFH(H3), que se asocia con menores niveles de FH y mayores niveles de FHR-325, del haplotipo CFH(H4), que se asocia con mayores niveles de esta proteína46, o de la deleción ΔCFHR3-CFHR1, que genera deficiencia de FHR-1 y FHR-3.
Por otro lado, varios estudios en pacientes de SHUa y NIgA sugieren que los niveles de FH y proteínas FHRs cambian también durante el curso de la enfermedad. Así, los niveles de FHR-1 y FHR-5, y las ratios FHR-1/FH y FHR-5/FH, son mayores en pacientes de NIgA que en las correspondientes poblaciones control, y hay una correlación directa entre los niveles de FHR-5 y la progresión de la enfermedad21,34,45. En el mismo sentido, algunos pacientes de NIgA tienen un mayor depósito de FHR-5 en los glomérulos, que correlaciona con el daño histológico y sirve de marcador pronóstico47. También se han observado cambios en los niveles de FHR-5 en algunos pacientes de SHUa asociado a infección con Streptoccus pneumoniae, que presentan niveles mayores de esta proteína en las muestras de plasma obtenidas en los primeros días del proceso infeccioso que en las muestras en remisión48. Esta probable influencia de la evolución clínica en los niveles de FH y proteínas FHRs explicaría también por qué los niveles de FHR-3 en pacientes de la cohorte española de SHUa son mayores que en los individuos sanos que tienen el mismo genotipo de CFHR327. Para abordar estos interrogantes, habrá que comparar los niveles de FH y proteínas FHRs en muestras seriadas de los pacientes, obtenidas al debut y en remisión. No hay publicaciones que documenten cambios en los niveles de FH y proteínas FHRs en pacientes de GC3; no obstante, resultados preliminares de nuestro grupo muestran que la mayoría de los pacientes tienen niveles elevados de algunas proteínas FHRs, y probablemente esta situación se da también en otras cohortes de GC3.
Otra cuestión que nos parece relevante es que en la mayoría de los estudios realizados en controles y pacientes se han comparado los niveles absolutos de FH y proteínas FHRs, pero sólo en algunos se han calculado los niveles relativos de las mismas, como las ratios FHR-1/FH o FHR-5/FH. El aumento en los niveles de una determinada proteína FHR puede ser relevante en un individuo con niveles constitutivamente bajos de FH, mientras que en un individuo con una concentración elevada de FH ese mismo aumento puede no ser significativo. En nuestra opinión, los cambios en las concentraciones relativas de FH y proteínas FHRs pueden ser tanto o más informativos que los cambios en los niveles absolutos de las mismas.
En resumen, aunque la cuantificación de las proteínas FHRs no se ha podido llevar a cabo de forma fiable hasta hace unos años, se sabe que los niveles de algunas de ellas, en particular FHR-1 y FHR-3, están determinados en gran medida por polimorfismos en la región CFH-CFHRs. Por otro lado, los datos obtenidos en pacientes de SHUa y NIgA revelan que los niveles de algunas proteínas FHRs y la ratio FH/FHRs se alteran durante el curso clínico de la enfermedad. Estos datos sugieren una contribución relevante de los niveles de FH y FHRs en la patogenia de enfermedades relacionadas con la desregulación de la VA del Complemento, que se irá perfilando a medida que mejore nuestra comprensión de la actividad funcional de estas proteínas y de los mecanismos que determinan sus niveles de expresión.
- •
El Factor H (FH) regula la vía alternativa del Complemento y evita que se active inespecíficamente sobre nuestras propias células y tejidos.
- •
Las proteínas FHRs son muy similares estructuralmente al FH, pero es probable que se comporten como antagonistas funcionales del FH y potencien la activación del Complemento.
- •
Los niveles de FH y proteínas FHRs están determinados parcialmente por polimorfismos en la región CFH-CFHRs cuya frecuencia varía entre poblaciones diferentes.
- •
Pequeños cambios en los niveles de FH y proteínas FHRs pueden desequilibrar la regulación del Complemento a nivel local o sistémico. y favorecer el desarrollo de patologías renales como el SHUa, las GC3 o la NIgA.
IGD tiene un contrato financiado por la Comunidad Autónoma de Madrid (Complement II-CM; S2017/BMD-3673) y por la Fundación Senefro (http://www.senefro.org/; Ayudas de Investigación 2018). El trabajo ha sido financiado por el Instituto de Salud Carlos III (ISCIII) y el Fondo Europeo de Desarrollo Regional (proyecto PI19/00970 a PS-C).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos a la Dra. Margarita López Trascasa y al Dr. Fernando Corvillo, miembros del Grupo de Investigación en Complemento del IdiPAZ, la lectura del manuscrito y sus comentarios sobre el mismo.



