





Gitelman's syndrome (GS) is an autosomal recessive disorder caused by mutations in the SLC12A3 gene. GS is characterized by hypokalaemic metabolic alkalosis, hypomagnesemia and hypocalciuria. Most of the reported patients of Roma ancestry are homozygous for an SLC12A3 intron 9 frameshifting mutation (c.1180+1G>T). Some forms of Bartter's syndrome result from mutations in the CLNCKB gene and clinically overlap with GS.
ObjectivesTo characterize a second SLC12A3 mutation in Roma patients negative for the intron 9 variant.
MethodsSLC12A3 and CLNCKB genes were analyzed by next-generation sequencing in two Spanish and Greek gypsy patients who were negative for the intron 9 splicing mutation. Sanger sequencing was performed to confirm the putative mutations in patients and family members.
ResultsWe identified a missense variant (p.Val647Met, c.1939G>A) in both cases, and both were homozygous for Met. This mutation was also found in three additional patients; two homozygous and one heterozygous compound with the intron 9 splicing mutation. This new SLC12A3 mutation seems to be characteristic of gipsy GS patients and was linked to the same haplotype in all cases, supporting a founder origin. All the patients showed biochemical features characteristic of GS.
ConclusionWe report a second founder mutation among GS patients of Roma ethnic background. The direct screening of this mutation would facilitate the characterization of patients who are negative for the more common intron 9 +1G>T mutation.
El síndrome de Gitelman (SG) es un trastorno autosómico recesivo causado por las mutaciones en el gen SLC12A3. El SG se caracteriza por una alcalosis metabólica hipopotasémica, hipomagnesemia e hipocalciuria. La mayoría de los pacientes de etnia gitana notificados son homocigotos para la mutación con desplazamiento del marco de lectura del intrón 9 de SLC12A3 (c.1180+1G>T). Algunas formas del síndrome de Bartter proceden de las mutaciones del gen CLNCKB y se solapan clínicamente con el SG.
ObjetivosDeterminar las características de una segunda mutación en SLC12A3 en pacientes de etnia gitana con resultados negativos en la variante intrón 9.
MétodosSe analizaron los genes SLC12A3 y CLNCKB mediante secuenciación de nueva generación en 2 pacientes –uno español y otro griego– de etnia gitana con resultados negativos en la mutación de empalme del intrón 9. Se llevó a cabo una secuenciación de Sanger para confirmar las supuestas mutaciones en los pacientes y sus familiares.
ResultadosSe identificó una variante con cambio de sentido (p.Val647Met, c.1939G>A) en ambos casos, y ambos eran homocigotos con respecto a Met. También se observó esta mutación en 3 pacientes adicionales, 2 homocigotos y uno heterocigoto compuesto con la mutación del intrón 9. Esta nueva mutación del SLC12A3 parece ser característica de los pacientes con SG de etnia gitana y se relacionó con el mismo haplotipo en todos los casos, lo que indica un origen fundador. Todos los pacientes presentaron rasgos bioquímicos propios del SG.
ConclusiónInformamos de una segunda mutación fundadora en los pacientes con SG de etnia gitana. El cribado genético directo de esta mutación facilitará la determinación de las características de los pacientes con resultados negativos en la mutación del intrón 9+1G>T, que es más frecuente.
Gitelman's Syndrome (GS) is the most common primary renal tubular disorder, affecting approximately 1/40,000 Caucasians. GS is inherited as an autosomal recessive disease mainly linked to mutations in the SLC12A3 gene (solute carrier family 12 sodium/chloride transporters, member 3), that encodes a thiazide-sensitive sodium-chloride co-transporter (NCC) protein that locates at the luminal membrane of the distal convoluted tubule in the kidney.1 GS is characterized by hypokalemic metabolic alkalosis with hypomagnesemia and hypocalciuria.2,3 The loss of function of the NCC channel leads to decreased sodium re-absorption and subsequently secondary volume depletion. The hypovolemia stimulates sodium re-absorption at the expense of increased potassium and hydrogen secretion, which results in hypokalemia and metabolic alkalosis.4 Hypocalciuria and hypomagnesaemia are also characteristic of GS, the latter being a likely consequence of the elevation of the excretion of magnesium in urine.5,6
Most of the GS patients are homozygous or heterozygous compound for rare private mutations. Due to the absence of common mutations, the sequencing of the full SLC12A3 is required to uncover the genetic background of GS patients.7 Moreover, some cases presenting as GS might carry mutations at the CLCNKB gene, a fact that increases the technical effort of the genetic study.7
The Roma or Gypsies represent the largest and most widespread ethnic minority in Europe. Anthropological and genetic data support their origin in the Indian subcontinent.8 Although there was some mixture with their hosting populations, consanguinity is common among Gypsies and results in the presence of founder-mutations for several inherited disorders.9 To date, most of the reported Gypsy patients having GS are homozygous for the intron 9 +1G>T mutation (c.1180+1G>T).10–12 Here, we describe a new SLC12A3 mutation characteristic of Roma SG patients.
MethodsPatientsWe studied eight patients from five families diagnosed of GS (Table 1). The five families had a Roma ethnic background and traced their ancestry to the South-East European region, including the Spanish family whose ancestors emigrated from Romania two generations ago. The Spanish and Greek families were recruited through the Renaltube Consortium (www.renaltube.com).13
Serum biochemistry and genetic data of the affected members in the five families.
| Kindred | Sex | Age at diagnosis | pH | HCO3− (mEq/L) | K+ (mEq/L) | Mg2+ (mg/dL) | Mutations in SLC12A3 | |
|---|---|---|---|---|---|---|---|---|
| PI.1 | Male | 14 years | 7.61 | 30 | 2.1 | 1.40 | p.Val647Met/p.Val647Met | |
| Family I | PI.1.1 | Female | 11 months | 7.50 | 28 | 3.9 | 1.50 | p.Val647Met/p.Val647Met |
| PI.1.2 | Female | 7 months | 7.50 | NA | NA | 1.83 | p.Val647Met/p.Val647Met | |
| PI.1.3 | Male | 11 months | 7.44 | 24.5 | 3.72 | 2.10 | p.Val647Met/p.Val647Met | |
| Family II | PII.1 | Female | 29 months | 7.48 | 28 | 2.5 | 1.80 | p.Val647Met/p.Val647Met |
| Family III | PIII.1 | Male | 7 years | 7.51 | NA | 1.8 | 1.00 | p.Val647Met/p.Val647Met |
| Family IV | PIV.1 | Male | 49 years | NA | 30.6 | 2.4 | 1.58 | p.Val647Met/p.Val647Met |
| Family V | PV.1 | Male | 33 years | NA | 34 | 2.6 | 0.96 | p.Val647Met/Intron 9 +1 |
K+: potassium; Mg2+: magnesium; HCO3−: bicarbonate; Val: valine; Met: methionine. NA: data not available.
The search for SLC12A3 and CLCNKB mutations was first performed in the index cases from families I and II. The two were negative for the intron 9 splicing mutation. The whole coding plus at least 5 intronic flanking nucleotides were sequenced through semiconductor Next Generation Sequencing (NGS) with the Ion Torrent Personal Genome Machine (PGM, Life Technologies), following a previously reported procedure.14
To confirm the putative mutations identified by NGS, the corresponding exons were amplified and sequenced by the Sanger method. This procedure was also used to determine the presence of the mutation in all the family members.
To characterize the haplotypes linked to the SLC12A3 mutation, five common single nucleotide polymorphisms were determined by Sanger sequencing of PCR fragments: rs5801 (c.1395C>T), rs2304483 (c.1670−8T>C), rs13306676 (c.2179−8C>T), rs34772420 (c.2748−13T>C), and rs2289115 (c.2951+13C>T). In families I and II we also genotyped an intron 9 repeat polymorphism.
ResultsClinical dataFamily I had several GS patients in two generations. The index case (PI.1) was the father of three children with mild clinical features of GS. The father was diagnosed in the adolescence whereas the three siblings were diagnosed before the age of two years. Three brothers and one sister of the index case presented also GS findings such as hypokalemia, hypomagnesaemia and elevated bicarbonate serum values with a blood pH>7.35 (data not showed).
GenotypeThe NGS of SLC12A3 and CLCNKB in the index cases from families I and II revealed the presence of a nucleotide variant in exon 16 of SLC12A3 (c.1939G>A) that would result in a missense amino acid change (p.Val647Met). This variant is predicted to be pathogenic/deleterious by the bioinformatic programs SIFT (http://sift.jcvi.org) and Polyphen (http://genetics.bwh.harvard.edu/pph2). The Sanger sequencing of exon 16 fragments showed that these patients were homozygous for the mutation, and we also determined the mutation genotype in all the family members (Fig. 1).
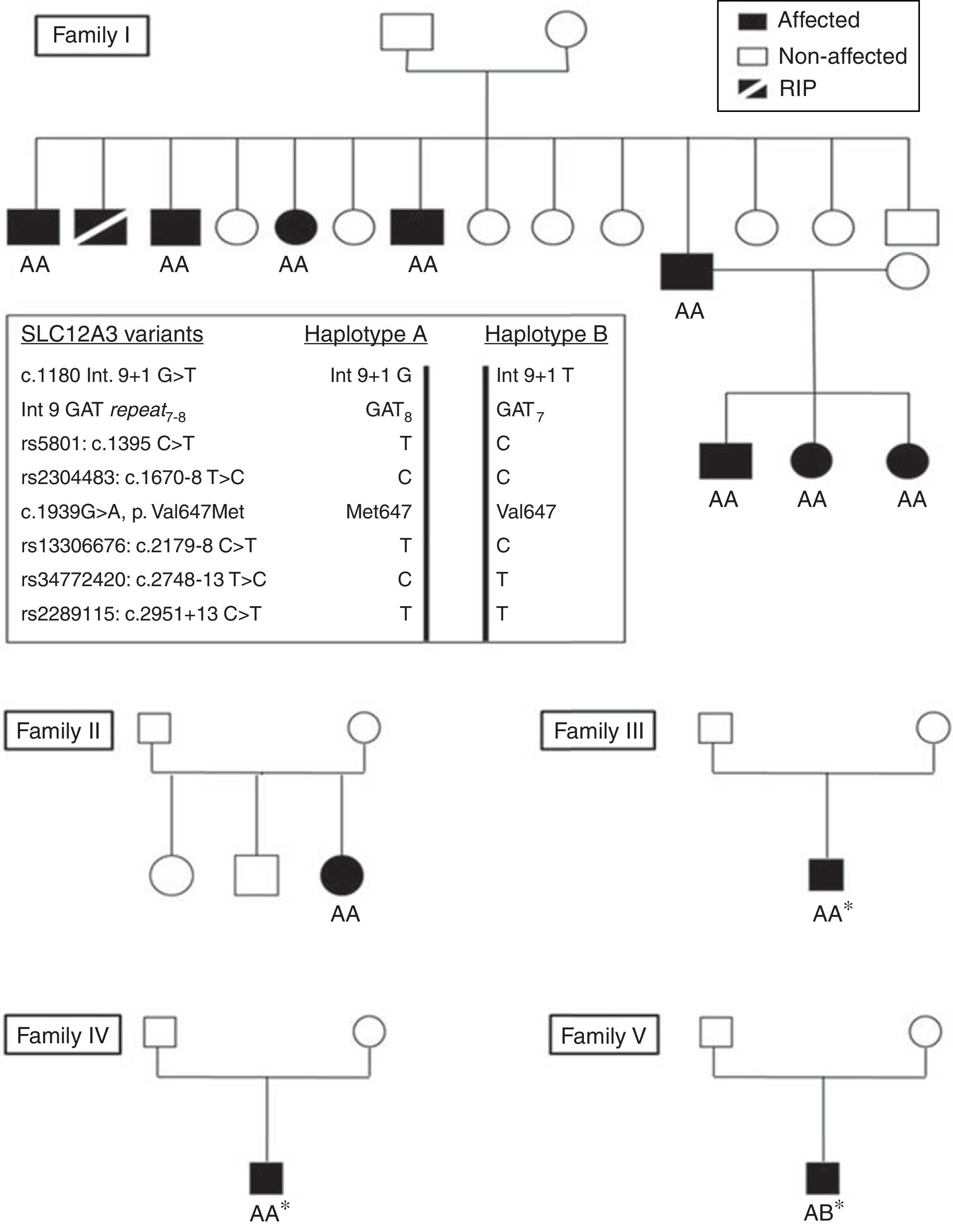
The analysis of the five SLC12A3 SNPs showed that the p.647 Val mutation was in the same haplotype in the five families (Fig. 1).
DiscussionMore than 180 mutations in SLC12A3 gene have been reported in GS patients.7 There is not a prevalent mutation, and this means that the sequencing of the 26 SLC12A3 exons is required to search for mutations in most of the GS patients.15,16 Moreover, CLCNKB mutations may be found in some cases with clinical manifestations of GS, a fact that increases the complexity of the genetic study of GS. Thus, the discovery of mutations common in an ethnic group will facilitate the genetic analysis by limiting the sequencing to one or few exons.10
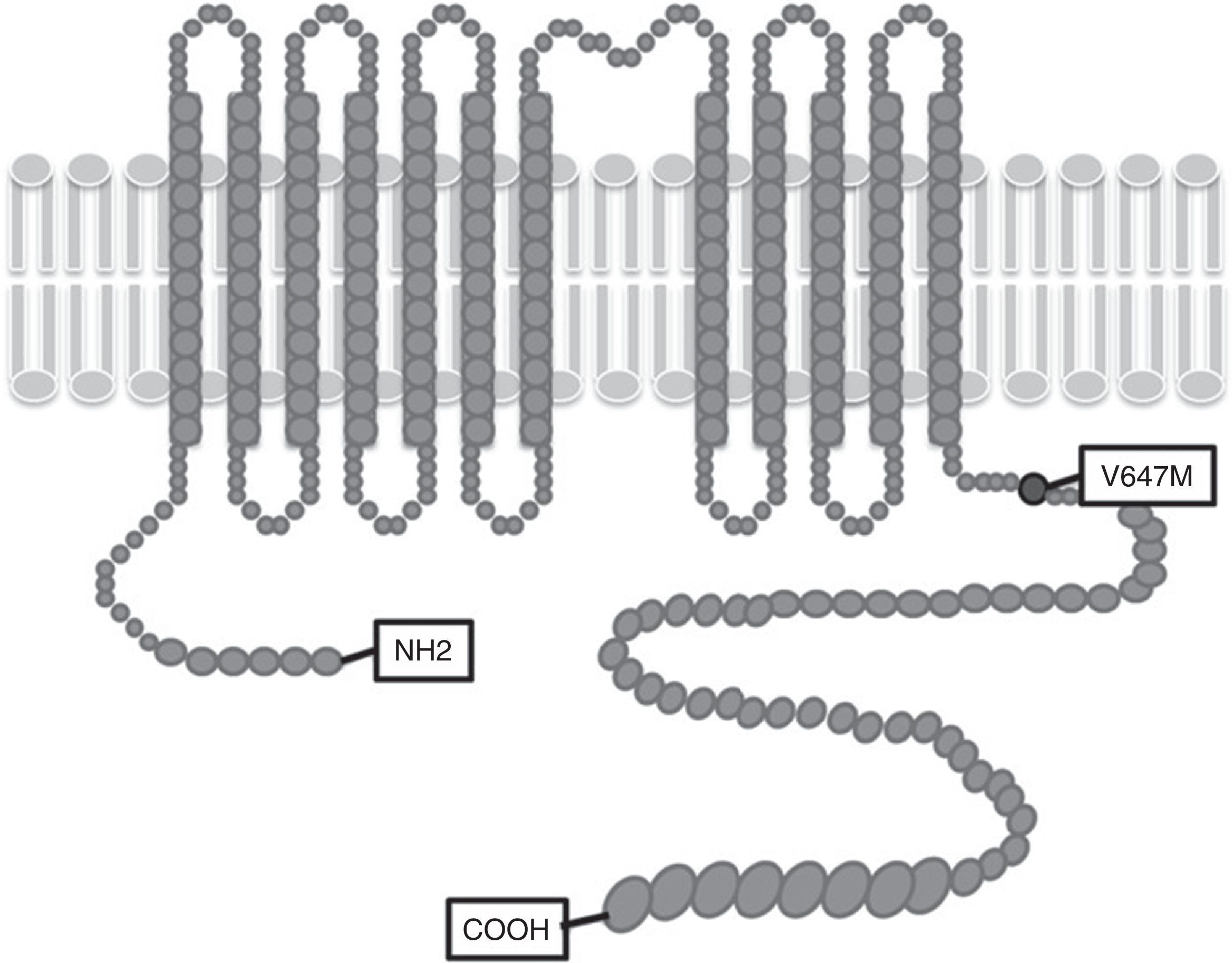
In this article we present Gypsy cases from Spain and Greece with the same mutation in the SLC12A3 gene. Patients were recruited through the Renaltube Consortium13 which collects data from patients worldwide in an attempt to better define the clinical profile and the genetic background of renal tubulopathies. To date, >50 Gypsi GS patients are included in the Renaltube database and most of these patients are homozygous for the intron 9 splicing mutation. The index cases of Families I and II, were subjected to NGS to search for new SLC12A3 mutations, resulting in the identification of p.Val647Met. The p.Val647Met variant was predicted to be pathogenic with at least two bioinformatic programs (SIFT and Polyphen). This missense change locates at the intracellular carboxy-end of the protein (Fig. 2), where other GS mutations have been found and aminoacids at this region are conserved not only among species but also in other human channels such us bumetanide-sensitive cotransporter (SLC12A2 gene).17
The p.Val647Met mutation had been previously reported by Vargas-Poussou et al. in one GS patient (PV.1) compound heteorzygous for this variant and the intron 9 mutation.7 Thus, the two families we studied and the three previously reported by Vargas et al.7 had a common Gipsy genetic background and traced their ancestry to South-Eastern Europe. The p.Val647Met was linked to the same SLC12A3 polymorphisms in the 5 families, a fact that strongly supports a common origin for this new Gipsy SG mutation.
In conclusion, we report a new SLC12A3 founder mutation among GS patients of Gypsy background, p.Val647Met. This mutation was associated with typical clinical features of GS, either in homozygosity or in combination with the intron 9+1G>A mutation. Our finding facilitates the diagnosis of GS in Gypsy patients.
Compliance with ethical standardsAll the patients/participants in the study gave their informed consent, approved by the Ethical Committee of Hospital Universitario Central Asturias.
Conflict of interestThe authors declare no conflict of interest.
This work has been supported by grant PI14/00702, Plan Nacional de I+D+I 2013–2016 and funded by ISCIII, Fondo Europeo de Desarrollo Regional (FEDER).



