




Anti-glomerular basement membrane (GBM) glomerulonephritis is an autoimmune disease triggered by autoantibodies against the NC1 domain of the α3 chain of type IV collagen (α3(IV)NC1). Anti-GBM glomerulonephritis rarely relapse after remission.1,2 Rare cases of anti-GBM glomerulonephritis complicated by IgG4-related disease (IgG4-RD) have been reported.3 Here, we report a rare case of anti-GBM glomerulonephritis complicated by type 1 autoimmune pancreatitis (AIP), a pancreatic manifestation of IgG4-RD, in a patient who experienced a relapse after remission.
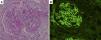
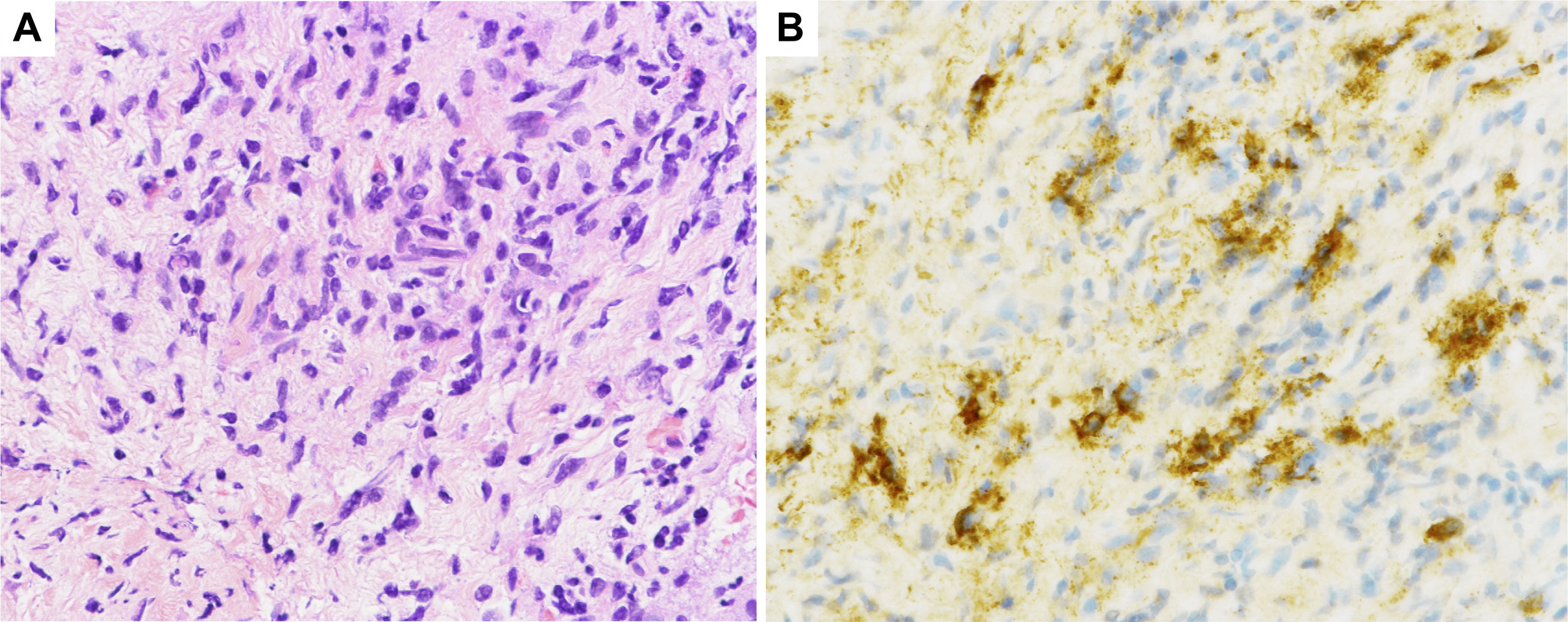
Case reportA 72-year-old man with fever and back pain was admitted to our institute. Three months before admission, he was diagnosed as having AIP based on the findings of pancreatic tail enlargement, irregular narrowing of the main pancreatic duct, and infiltration of lymphocytes and IgG4-positive plasma cells, together with storiform fibrosis by a pancreatic biopsy (Fig. 1), in accordance with the Japanese diagnostic criteria for AIP.4 He had been followed up without any medication. He had no history of smoking. His physical examination results were unremarkable except for a fever with a body temperature of 38.0°C. Laboratory studies showed worsening of his renal function (increased serum creatinine [Cr] level to 1.77mg/dL from a baseline of 0.5mg/dL), proteinuria (2.07g/gCr), hematuria (30–49 red blood cells per high-power field), and a C-reactive protein level of 30.9mg/dL. His serological test results were positive for anti-GBM antibodies (1500U/mL) but negative for anti-neutrophil cytoplasmic antibodies (ANCAs). Plain computed tomography revealed no signs of alveolar hemorrhage or acute pancreatitis. Renal biopsy revealed 8 glomeruli, all of which had a cellular crescent formation. No signs of IgG4-related tubulointerstitial nephritis (TIN) were found. Immunofluorescence microscopy revealed positive linear staining for IgG (IgG1>IgG4) and C3 along the GBM (Fig. 2). On the basis of these findings, the patient was diagnosed as having anti-GBM glomerulonephritis. He received intravenous pulse methylprednisolone (500mg daily for 3 days) followed by prednisolone (30mg daily) and plasma exchange (three times weekly for 25 occurrences). Five months later, his anti-GBM antibody level was undetectable, so the prednisolone dosage was tapered to 5mg daily with a stable serum Cr level of 2.42mg/dL. One year after treatment initiation, his serum Cr level increased to 7.40mg/dL and he tested positive for anti-GBM antibodies (681U/mL). He was diagnosed as having relapsed anti-GBM glomerulonephritis and underwent hemodialysis. He was treated with intravenous pulse methylprednisolone followed by prednisolone and plasma exchange. Nine weeks later, his anti-GBM antibody level was successfully reduced to 8.3U/mL, but he remained dialysis dependent.
Histological sections of pancreatic fine-needle aspiration specimens. (A) Hematoxylin and eosin staining showing storiform fibrosis (original magnification ×400). (B) IgG4-immunohistochemical staining showing a large number of IgG4-positive plasma cells (>10 per high-power field, original magnification ×400).

Histological sections of renal biopsy specimens. (A) Periodic acid-Schiff staining showing a cellular crescent formation (original magnification ×400). (B) Immunofluorescence staining showing linear patterns of IgG deposition along the glomerular basement membrane (original magnification ×400).
We report a rare case of relapsed anti-GBM glomerulonephritis complicated by AIP. Our case highlights two important clinical issues: anti-GBM glomerulonephritis can relapse after remission and can be complicated by IgG4-RD.
In typical anti-GBM disease, the risk of relapse is thought to be quite low (1.2–2.3%).1,2 Although a case of relapse triggered by smoking has been reported,5 our patient had no history of smoking. Recently, two atypical variants of anti-GBM disease have been reported. One is anti-GBM disease with double seropositivity, for ANCAs and anti-GBM antibodies, with a relapse risk of 22%.6 The other variant is anti-GBM disease with a predominance of subclass IgG4, with a relapse risk of 50%.7 We ruled out these atypical types.
To our knowledge, no previous reports have described cases of anti-GBM glomerulonephritis complicated by AIP. One report described two cases of anti-GBM disease complicated by IgG4-related TIN.3 Both cases presented with acute kidney injury and were diagnosed as anti-GBM glomerulonephritis and IgG4-related TIN based on the findings of seropositivity for anti-GBM antibodies and IgG linear deposition in the GBM and infiltration of IgG4-positive plasma cells by renal biopsy. Both cases were serologically double-positive for anti-GBM antibodies and ANCAs, and had a poor prognosis.
It is important to distinguish IgG4-RD from Castleman disease (CD) because both show plasma cell infiltration. CD is characterized by lymph node proliferation and systemic inflammation. A case series study reported three cases of anti-GBM disease complicated by CD.8 The authors detected plasma cells secreting anti-α3(IV)NC1 antibodies in lymph nodes. One of the three patients experienced a relapse. Therefore, they suggested a causal relationship between plasma cell infiltration and anti-GBM antibody production. In our case, plasma cell infiltration was observed in the pancreas; therefore, our patient may be at high risk of relapse and require careful follow-up.
In conclusion, anti-GBM glomerulonephritis can be complicated by IgG4-RD and relapse after remission.
Compliance with ethical standardsThe authors have no conflicts of interest to declare. This research did not receive any specific grant from funding agencies. All procedures performed were in accordance with the ethical standards of the institutional research committee (IRB approval number: 3-019068-00) and with the Helsinki declaration. Informed consent was obtained from the patient.
Conflict of interestNone.
We would like to thank Editage (www.editage.jp) for English language editing.



