


Vascular calcification has traditionally been considered to be a passive process that was associated with advanced age, atherosclerosis, uncommon genetic diseases and some metabolic alterations such as diabetes mellitus and end-stage kidney failure. However, in the last years, vascular calcification has been proven to be an active and regulated process, similar to bone mineralisation, in which different bone-related proteins are involved. Recent results question the classic classification of vascular calcification into intimal and medial calcification, at least in capacitance arteries. Pro and anti-calcifying mechanisms play an active role in calcium deposition in vascular cells, making this area an active focus of research. The identification of therapeutic targets which can slow down the progression or even reverse vascular calcification could be an important step forward in the treatment of patients with chronic kidney disease.
Clásicamente se consideraba que la calcificación vascular era un proceso pasivo y degenerativo que frecuentemente ocurría con la edad avanzada, aterosclerosis, varias alteraciones metabólicas (como diabetes mellitus y estados finales de enfermedad renal) y en raras enfermedades genéticas. Sin embargo, desde hace algunos años, la calcificación vascular es considerada como un proceso activo y regulado de manera semejante a la mineralización y metabolismo del hueso, en el se encuentran implicadas diversas proteínas óseas. Resultados recientes cuestionan la clásica separación de la calcificación vascular en calcificación de la íntima y calcificación de la media, al menos en arterias de capacitancia. Mecanismos procalcificantes y anticalcificantes desempeñan un papel activo en la deposición de calcio en las células vasculares, por lo que su estudio se ha convertido en un área muy activa de investigación. La identificación de dianas terapéuticas que puedan enlentecer o incluso revertir la calcificación vascular podría suponer un avance muy importante en las estrategias terapéuticas para los pacientes afectados de enfermedades renales.
INTRODUCTION
Calcium phosphate may be deposited as bioapatite crystals (similar to bone) in blood vessels and heart valves in vascular calcification.1 Traditionally, calcification has been classified depending on where the calcium was deposited. In this way, arterial calcification has been divided into intimal calcification (associated with atheromatous plaques2) and medial calcification (known as Mönckeberg’s sclerosis) linked to vascular stiffness due to the mineralisation of elastic fibres and atherosclerosis seen with age, diabetes and chronic kidney disease (CKD).3 The first one would be linked to an increased deposit of lipids and inflammatory cell infiltrate, while the phenotypic transformation of vascular smooth muscle cells towards osteoblast-like cells would be more important in the second one. A mixture of both calcifications is seen in patients with CKD.4,5 However, recent results seem to suggest that this classification is not very clear and that both would be manifestations of the atherosclerotic process,6 at least in great arteries. Mönckeberg first described this in 1903.7 He described in his article the presence of calcification in the middle layer of 18 patients’ arteries with no evidence of plaque. However, the description was made without the help of modern-day techniques to measure lipid deposit, extracellular matrix, etc. It would not be too far fetched to think that what he was actually describing was different stages in the evolution of atherosclerotic plaque. However, several studies have described patients with Mönckeberg’s sclerosis in the last few years.8-12 If we analyse these studies in detail, we can come to the conclusion that there are characteristics of atherosclerotic lesions in nearly all of them: increased intima-media thickness, disruption of the internal elastic lamina or even lipid deposits. Furthermore, the great arteries have a middle layer with a low number of vascular smooth muscle cells. They are, therefore, more sensitive to the atherosclerotic process than to phenotypic transformation towards osteoblast-like cells.
A recent study by our group using ultrasound, the only noninvasive method to determine the exact location of vascular calcifications, shows that vascular calcification of capacitance arteries is associated with the presence of atherosclerosis.13 In this paper, we have studied the presence of vascular calcifications and atheromatous plaques in carotid, femoral and brachial arteries in 232 patients and 208 control patients. The most common type of vascular calcification was linear calcification of the intima, followed by atheromatous plaque calcification. What seemed to be a new type of vascular calcification is not, in fact, since calcification of the internal elastic lamina had histologically already been described in coronary arteries.11 Another important result was that linear calcification of the intima was intimately associated with the presence of plaques, as it was not found in radial arteries (which do not develop atherosclerosis). The study also concluded that absence of carotid plaque was a protective factor. Therefore, our results seem to indicate that the predominant type of vascular calcification of great arteries in patients on dialysis is associated with the presence of atherosclerosis.
VASCULAR CALCIFICATION MECHANISMS
Vascular calcification is an active and regulated process that involves different mechanisms that are not mutually exclusive.14
Calcium and phosphorus
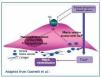
Some authors refer to them as “passive mechanisms of calcification”. Elevated levels of Ca, P and CaxP (prevalent in patients with CKD and significantly associated with death due to cardiovascular disease [CVD] in these patients15) cause clusters of bioapatite crystals to form and grow.16 Bioapatite is the main mineral component of bones, fish bones and shells. In vitro studies found that when VSMC were incubated with high concentrations of calcium or phosphorus, bioapatites accumulated in the extracellular matrix. When they were incubated with both elements at the same time, a synergistic effect of calcification was observed.17 However, this process is not just a passive precipitation of divalent ions, but rather a phenotypic change of VSMC and the up-regulation of genes commonly associated with bone differentiation.18 The effects of hyperphosphataemia are mediated by a sodium-dependent phosphate cotransporter (NPC). Type III NPC, Pit-1, has been found in VSMC. High phosphorus levels stimulate the load while elevated calcium levels increases the Pit-1 mRNA expression. This transporter allows phosphorus to accumulate within cells, which acts as a signal for the expression of osteogenic genes. This causes mineral molecules to be secreted (matrix vesicles, calcium-binding proteins, alkaline phosphatase and collagen-rich extracellular matrix). The combination of these factors induces the cell to change and become susceptible to calcification (Figure 1).
Cell death and apoptosis
Vascular calcification is linked to the appearance of matrix vesicles with cytoplasmic content and intact cell membrane (as happens in bone development). These vesicles are formed from cells where mineralisation starts or they are the result of the cell apoptosis process (apoptotic bodies). The wall of uraemic patients is damaged by inflammation processes and oxidative stress and one may therefore think that there is cell apoptosis. Proudfoot et al.19 showed that apoptosis regulates vascular calcification in vitro. According to these authors, matrix vesicles are capable of concentrating calcium inside and bioapatite crystals originate in them.
Calcification inhibitors
Under normal conditions blood vessel cells express mineralisation-inhibiting molecules. The loss of their expression, as happens in CKD, causes what is known as “loss of natural inhibition”, giving rise to spontaneous calcification and increased mortality. A list with these calcification-inhibiting molecules has been drawn up after mutation analysis on mice, including among others:
Matrix Gla Protein
Matrix Gla Protein (MGP) was the first calcification inhibitor to be identified. It is a vitamin K-dependant protein that is constitutively expressed in VSMC and endothelial cells of normal blood vessels, but its expression is greatly reduced in calcified arteries.20 It has also been observed that its expression is reduced in in vitro calcification models.21 Serum MGP levels are lower in patients with calcifications than in those without it.22 Furthermore, MGP knockout mice develop severe medial calcifications and die of a ruptured aorta.23
Fetuin A
Fetuin A is a serum glycoprotein that inhibits ectopic vascular calcification. It is a powerful inhibitor of hydroxyapatite formation, reducing the formation of crystals in in vitro solutions containing calcium and phosphorus without affecting those that are already formed.24 Mice that are deficient in this protein develop extensive calcifications in soft tissue such as the myocardium, kidneys, tongue and skin.25
Osteopontin
Osteopontin (OPN) is a phosphoprotein that is usually found in mineralised tissue such as bones and teeth, and is involved in regulating mineralisation as it inhibits apatite crystal growth.26 Although it is not found in normal arteries, some authors have detected its expression in atherosclerotic plaques and calcified aortic valves.27-29 Giachelli et al.30 crossed OPN-/- mutant mice (that had no vascular symptoms) with MGP-/- mutant mice (that had developed vascular calcifications) to examine the role of OPN in vascular calcification. OPN-/- MGP-/- mice showed a more accelerated calcification than those that were only deficient in MGP (MGP-/- OPN+/+). These studies, therefore, indicate that OPN is an inducible inhibitor of vascular calcification in vivo.
Osteoprotegerin
Osteoprotegerin (OPG) is a member of the tumour necrosis factor receptor family that has been identified as a regulator of bone resorption.31 OPG is produced by many tissues, including the cardiovascular system, lungs, kidney and immune system.32 In advanced calcified lesions, OPG is found around the calcified area. It has been seen that OPG-deficient mice develop severe osteoporosis and medial calcification.33 Therefore, OPG is obviously an inhibitor of vascular calcification. The potential of OPG as a marker of cardiovascular disease has been studied. As the severity of vascular calcification increases so does the serum OPG level.34 OPG functions as a soluble decoy receptor for the receptor activator of NF-kB (RANK) ligand (RANKL).32 RANKL is produced by activated T cells and stimulates RANK. This activation enables, among others, an increased expression of inflammation mediators. In addition, OPG is a receptor for tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), which is a powerful apoptosis inducer. TRAIL is found in many different tissues, including VSMC and endothelial cells. In human atherosclerotic lesions, TRAIL has been located around calcified areas.9
Calcification activators
There are studies that speculate that, as well as hyperphosphataemia and hypercalcaemia, there are substances present in the blood serum of patients with CKD capable of stimulating calcification.35 Bovine VSMC in the presence of uraemic serum increases the expression of calcification-related proteins. A large number of uraemic factors have been identified that are capable of inducing osteogenic genes, transforming osteoblasts and secreting some bone matrix proteins in the walls of blood vessels and soft tissue. Some of these factors are: tumour necrosis factor (TNF),36 inflammatory cytokines,37 fibronectin,38 type-I collagen38 and 25-hydroxycholesterol.39 These uraemic serum substances stimulate the expression of molecules essential to vesicular calcification.
Alkaline phosphatase
Alkaline phosphatase (ALP) is one of the osteoblastic phenotype markers and is considered essential in the vascular calcification process. It has been detected in vascular and heart valve calcifications. ALP expressed on the surface of cells can act on phosphate liberators, releasing inorganic phosphate.40 Inflammatory cytokines and vitamin D induce its up-regulation and mineralization.40,41
Core-binding factor alpha 1
Core-binding factor alpha 1 (Cbfa1) is the main regulator of bone cell differentiation. Cbfa1-deficient mice have problems with cartilage formation and bone mineralisation.42 It acts as a transcription factor that accelerates the expression of important osteoblast lineage genes such as osteocalcin, osteopontin, ALP or type-I collagen.20 Its expression is up-regulated by phosphate43 and uraemic toxins.35
Bone morphogenic proteins
Bone morphogenic proteins (BMP) are a group of, at least, 30 proteins that receive their name from their osteoinductive properties. BMP belong to the transforming growth factor-beta (TGF-‚) superfamily. They act by binding to a heterodimeric system of transmembrane receptors (BMP-1 and BMP-2 receptor) that trimerises upon binding. The binding of a BMP to its specific type II receptor results in the type 1 receptor being activated. This causes phosphorylation and nuclear translocation of the Smad transcription factors thus modifying the transcription rate of target genes.44 They then induce ectopic bone formation.45
BMP2 is a powerful bone morphogenic protein and its expression triggers osteogenic transcriptional regulatory programs in the arterial tree. BMP2 induces Msx2 as well as Cbfa1 in VSMC.46 Msx2 is needed for the formation of intramembranous bones and it is critical for osteoblast differentiation, endochondral bone formation and neovascularisation.
They were recognised as mediators of vascular calcification long ago: BMP2 and BMP4 are involved in mineralisation and induction of local inflammation, while BMP7 slows down vascular calcification. BMPs are expressed in different cells in atherosclerotic lesions as well as in endothelial lesions and VSMC.47,48 The effect of BMP2 on vascular calcification is inhibited by MGP.49
RANKL
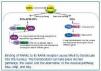
RANKL (also known as OPGL) is a protein consisting of 316 amino acids with a molecular weight of 38kD. Its expression is also modulated by several cytokines, glucocorticoids and PTH.50 RANKL is produced by osteoblast lineage cells and activated T cells. It promotes osteoclast formation, fusion, differentiation, activation and survival, leading to increased bone resorption and bone loss.51 RANKL stimulates its specific receptor RANK, which is expressed in fewer cells such as progenitor cells and mature osteoclasts, activated T cells and dendritic cells.52-54 The activation of RANK by RANKL triggers the NF-κB intracellular signalling cascade. The final stage of RANK activation is the NK-κB translocation into the nucleus, which can take place by the classical or alternative pathway. Both pathways are regulated by their kinases which are, respectively, IKK‚ and IKKα. The NK-κB translocation to the nucleus modulates the expression of different genes, e.g. BMP4 (Figure 2).55
The biological effects of OPG are the opposite of RANKL-mediated effects, due to the fact that OPG acts as a soluble inhibitor that prevents RANKL interaction and the subsequent stimulation of its RANK receptor.56
The first signs that this system was involved in vascular calcification came out of a study on OPG-knockout mice, which had osteoporosis and calcifications of the aorta and kidney arteries.33 OPG expression can be found in the media of great arteries31 and in many different types of blood vessel cells, such as VSMC and endothelial cells.57,58 It has been proven that it acts as an autocrine survival factor in endothelial cells.58 In contrast, RANKL and RANK have only been found in calcified areas of transgenic mice, in the arteries of wild mice.59 Other studies have demonstrated that OPG inhibits vascular calcification in in vivo rats caused by both vitamin D and warfarin.60 The definitive proof that RANKL directly promotes vascular calcification came in 2009, when one of the studies from our laboratory proved that RANKL directly increases calcification of VSMC by increasing BMP4 expression. This increased expression is due to the activation of the alternative NFkB signalling pathway.
KEY CONCEPTS
1. Recent results seem to indicate that vascular calcification is always associated with the presence of atheromatous plaques in great arteries, more than with mineral metabolism disorders. This does not rule out that mineral metabolism disorders might intensify vascular calcification.
2. Pro-calcifying and anti-calcifying mechanisms play an important role in the pathophysiology of vascular calcification. Therapies that aim to reduce vascular calcification in patients on dialysis should be directed at trying to reduce atherosclerosis as well as restoring anti-calcifying mechanisms or inhibiting pro-calcifying mechanisms.

Figure 1. Model of the effects of calcium and phosphorus on the mineralisation of VSMC.

Figure 2. Diagram of the activation of RANK by RANKL.



