



Hereditary kidney diseases (HKDs) are a heterogeneous group of kidney diseases that account for 10% to 15% of patients who start renal replacement therapy (RRT), and could account for an even greater percentage in the paediatric population.1 HKDs require a diagnostic approach that is different from other nephropaties since its pathogenesis is due to a mutation. The diagnosis goes beyond the affected patients, it includes the family. Ait is necessary to construct a family tree including both affected and non affected individuals.
Autosomal dominant polycystic kidney disease (ADPKD), which is due to a mutation in the PKD1 or PKD22 gene, is the most common hereditary kidney disease and accounts for 6–10% of patients in RRT. It is characterised by the growth of kidney cysts that lead to a decline in kidney function until RRT is required.3 In addition, it is associated with other manifestations: hypertension (HTN),4 polycystic liver disease, brain aneurysms and valve diseases.5 Alport syndrome (AS) is classified according to its pattern of inheritance: 85% X-linked owing to a mutation in COL4A56 and 15% autosomal recessive owing to a mutation in COL4A3 or COL4A4.7 In AS, an abnormal glomerular basement membrane causes microhaematuria, proteinuria and progressive CKD, which may be associated with eye abnormalities and hearing loss.8 Benign familial microhaematuria (BFM), which may affect up to 1% of the population, and recessive AS have a common pathogenesis.9 Tubulopathies are another group of HKDs, by which the mutation of a protein involved in tubular reabsorption fosters the excretion of certain ions in the urine.10 In addition, there are other hereditary syndromes with kidney disease in a context of significant multi-organ involvement such as tuberous sclerosis11 and Bardet–Biedl syndrome.12
Our objective was to create a specific practice for the follow-up of patients with HKD. These diseases tend not to be very prevalent, and so it is necessary to group them in a practice followed up by the same physician to increase his or her clinical experience. Below we present the steps that we followed, the difficulties that arose and our results after 3 years of follow-up.
Study design and patient screeningIn February 2012, a specific practice was created for the follow-up of patients with HKD in our department, which covers a population of nearly 300,000 inhabitants and has approximately 1500 outpatient visits per year. Our hospital is a tertiary hospital. Patients with kidney disease and a prior family history with a high clinical probability of HKD or with a positive genetic test were enrolled. Patients with a glomerular filtration rate lower than 25ml/min were excluded.
Patients were referred from Primary Care, Paediatric Nephrology and other Nephrology practices and by other specialists to an HKD-specific practice followed up by the same nephrologist. They were discharged from this practice if HKD was ruled out, and relatives with kidney diseases were enrolled after careful screening was performed. Follow-up lasted 3 years. The study ended in January 2015 (Fig. 1).
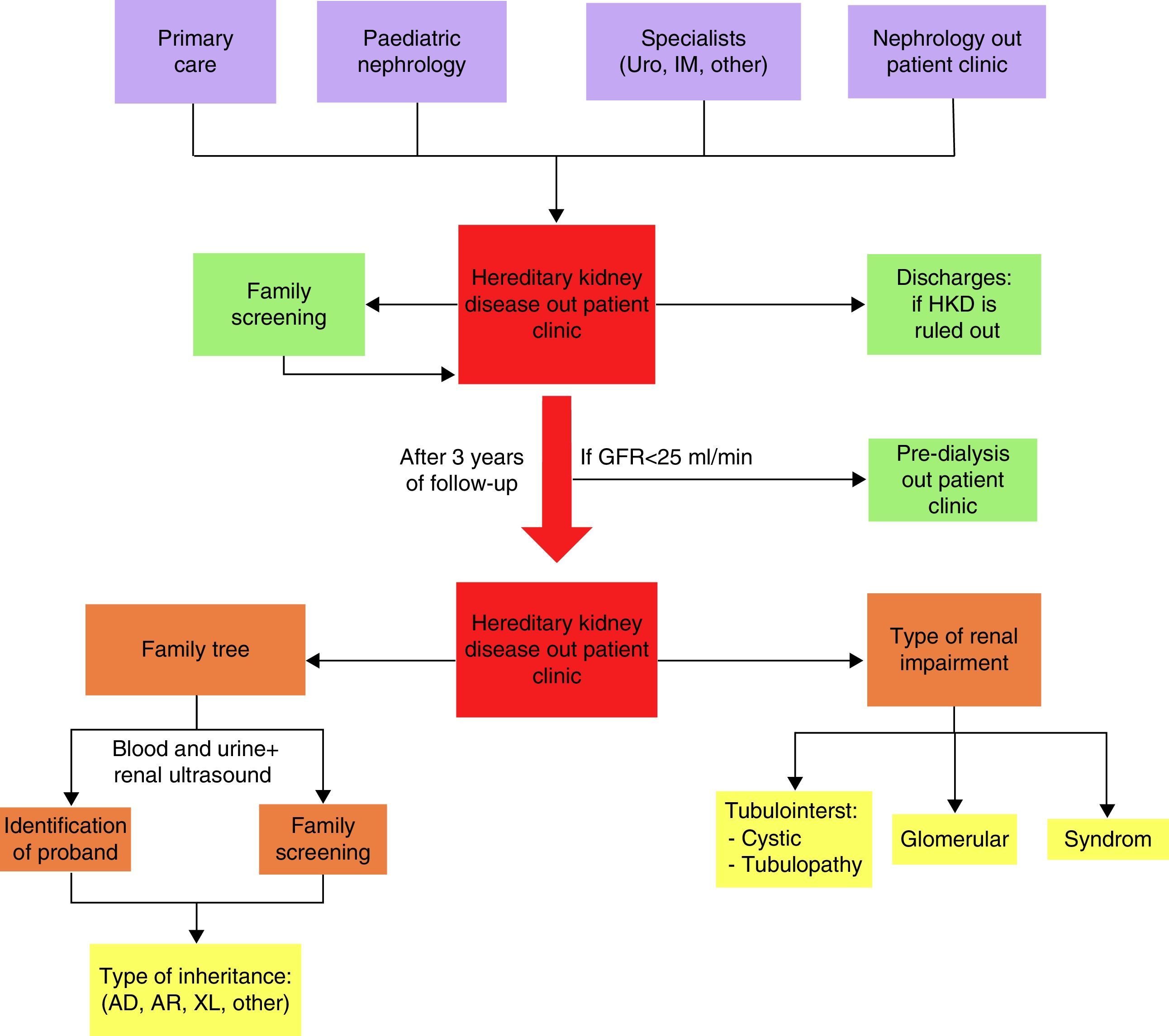
All the patients underwent blood and urine testing, a kidney ultrasound, measurement of blood pressure and preparation of a family tree that gathered family relationships with proband, healthy relatives and relatives with kidney disease (microhaematuria, proteinuria, kidney cysts, chronic kidney disease and requirement for RRT). With these data, 3 basic clinical patterns were established: tubulointerstitial, subdivided into cystic (cystic diseases by ultrasound diagnosis) and tubulopathic (ion abnormalities due to increase in urinary excretion); glomerular (proteinuria and microhaematuria); and syndromic (kidney disease in a context of significant multi-organ abnormality). The clinical patterns, together with the pattern of inheritance (autosomal dominant [AD], autosomal recessive [AR] and linked to the X chromosome [XL]), guided the physician towards an initial suspected clinical diagnosis in each patient (Fig. 1).
At the end of the study, the following data were collected: age, gender, performance of a kidney biopsy or genetic study, departments having referred the patient to the HKD practice, existence of a prior diagnosis, follow-up by other specialists owing to his or her HKD, performance of complete family screening, HTN, treatment with renin–angiotensin system inhibitors, diabetes mellitus, dyslipidaemia and whether the subject was a smoker.
In ADPKD, there was no radiologist assigned specifically to analyse ultrasound data: kidney size measured in centimetres, description of normal or large kidneys and presence of liver cysts. Familial microhaematuria (FM) was defined as microhaematuria in the patient and at least one of his or her relatives, other causes having been ruled out with immunology, serology and lithiasis studies. The following were included within FM: AS by consistent genetic or histological diagnosis and familial IgA nephropathy by kidney biopsy with a positive family history. In this group, the hearing tests performed were assessed by the otorhinolaryngologist as normal or with hearing loss.
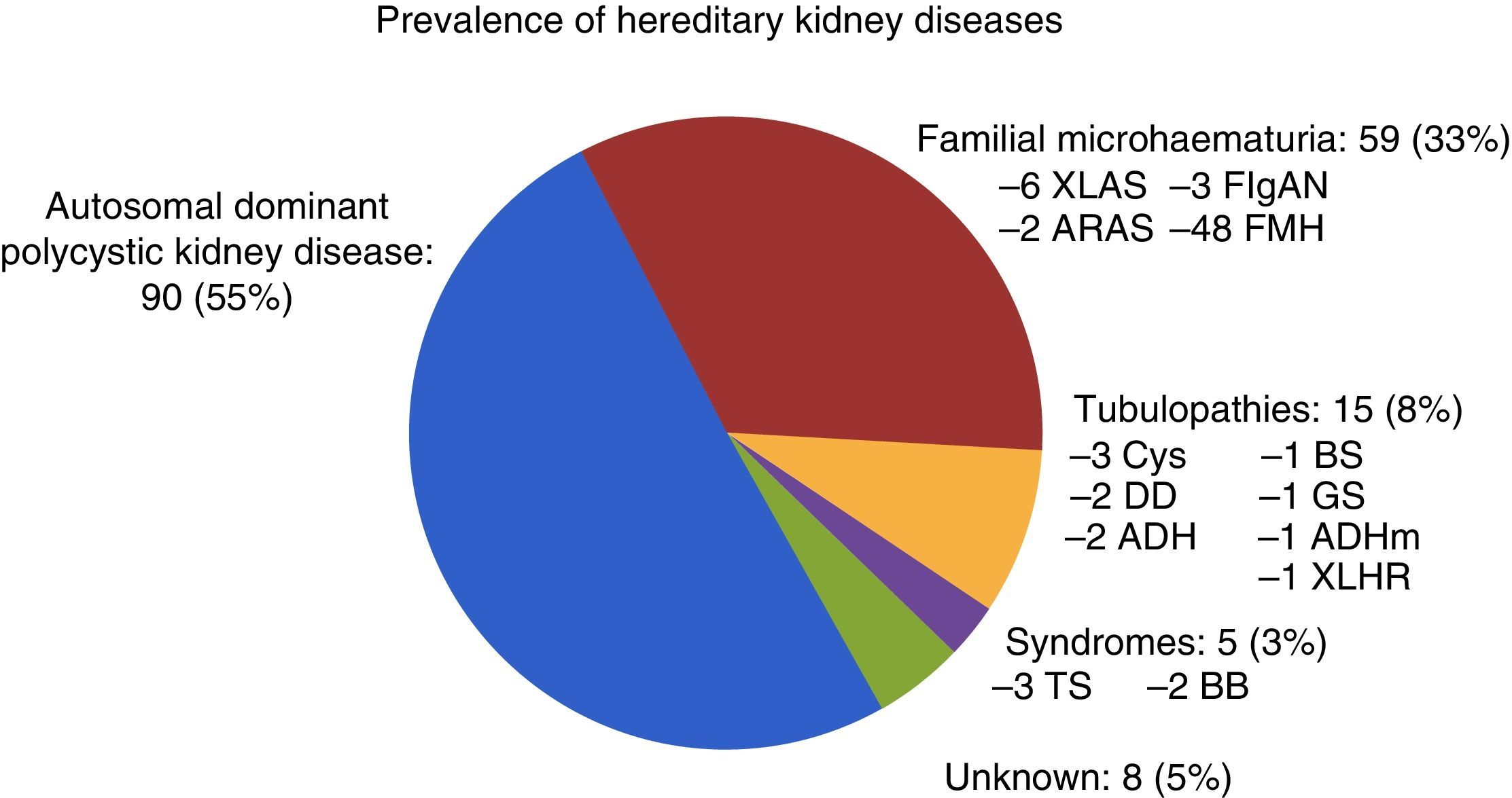
Of the 177 patients followed up in the HKD practice, 73 were men (41.2%) and 104 were women (58.8%). The mean age was: 43.13±15.20 (14–84) years. A family tree was prepared for all the patients; family screening was complete in 81 patients (45.8%) and incomplete in 96 patients (54.2%) owing to their relatives’ death or refusal. A total of 117 patients (66.1%) had at least one relative in RRT. Regarding patterns of inheritance: AD in 143 (80.1%), AR in 16 (9%), XL in 11 (6.2%) and unknown in 7 (4%). A genetic study was requested in 34 cases and positive in 21 (61.8%). Of the 8 kidney biopsies performed, only 3 were diagnostic for IgA nephropathy, and the rest were normal. Fig. 2 shows the prevalence of HKD in our practice.
The origin of the patients followed up in the HKD practice was: 61 (34.5%) from Primary Care; 14 (7.9%) from Paediatric Nephrology; 51 (28.8%) from other Nephrology practices; 17 (9.6%) from other specialists, mainly Urology and Internal Medicine; and 34 (19.2%) from family screening performed in the HKD practice. Only 73 patients (41.2%) were diagnosed before being referred to our practice.
A total of 49 patients (27.7%) were followed up by other specialists owing to abnormalities deriving from their HKD: 19 by Urology, owing to lithiasis, repeated urinary infections or complicated cysts; 11 by Otorhinolaryngology owing to hearing loss; 5 by Cardiology owing to ischaemic cardiomyopathy or mitral valve insufficiency; and 4 by Gastroenterology owing to massive polycystic liver disease or diverticulosis.
An analysis of cardiovascular risk factors revealed that 94 patients (53.1%) were hypertensive, 15 (8.5%) were diabetic, 58 (32.8%) had dyslipidaemia and 42 (23.7%) were smokers. In addition, 104 patients (58.8%) were treated with renin–angiotensin system inhibitors.
The ultrasound data in the ADPKD group were as follows: liver cysts were present in 49 patients (54.44%) and kidney size was only measured in 45 (50%), although in all patients it was determined whether their kidneys were normal or large. Hearing tests were performed in the group with FM in 45 patients (75%), 26 of whom had hearing loss and only 13 of whom were informed by the otorhinolaryngologist.
The majority of patients were referred to our practice owing to a suspicion of HKD, as they had kidney disease and at least one relative in RRT, although nearly 60% were not initially diagnosed. To establish a clinical diagnosis, our main tool was not the genetic study, but the family tree. In addition to examining the patient, it is essential to perform comprehensive family screening, with laboratory testing and a kidney ultrasound, distinguishing between healthy individuals and individuals with kidney disease to identify the type of inheritance and clinical pattern. However, in half of the cases family screening was incomplete owing to some family members’ refusal or death.
Proper diagnosis of HKD is important for managing factors for progression, establishing a multidisciplinary approach, performing suitable recording, offering genetic counselling and, in some cases, starting the appropriate treatment, such as enzyme replacement in Fabry disease13 or everolimus in tuberous sclerosis.14 However, there are familial kidney diseases of unknown origin or with a very atypical clinical presentation that are the subject of research studies, where collaboration between hospitals is required to create a national network that includes more patients with these low-prevalence diseases. The latest next-generation genomic sequencing techniques are contributing to the evolution and optimisation of HKD diagnosis.15
ADPKD is the most common hereditary kidney disease and our data is consistent with this. Ultrasound is the technique of choice for ADPKD diagnosis, in conjunction with follow-up and screening in first-degree relatives.16 Kidney volume is the best predictor of disease progression.17 However, in half of the cases in our radiological study, kidney size was not assessed precisely. In addition, an ultrasound must be performed to diagnose liver cysts, the most common extrarenal manifestation of ADPKD.2 It is essential that awareness be raised and that imaging tests be performed by a single physician trained in these diseases.
Clinically, it is difficult to distinguish between BFM, AS carrier status and familial IgA, since they may have the same glomerular pattern.18 Although in our study they were grouped together as FM, a definitive diagnosis should be made. The first step in making a differential diagnosis is determining the pattern of inheritance: AD in BFM, AR or XL in AS and polygenic inheritance in familial IgA (although the genes involved are being researched).19 A definitive diagnosis of AS is made by finding a dysfunctional glomerular basement membrane in electron microscopy or a pathogenic mutation in COL4A5 in XL Alport syndrome, or 2 pathogenic mutations in COL4A3 or COL4A4 in AR Alport syndrome, while in BFM there would only be a mutation in COL4A3/COL4A4.9
There are characteristic multi-organ abnormalities associated with certain HKDs that may help to identify them. It is essential to take a multidisciplinary approach. In many cases, the nephrologist becomes the coordinator of the other specialties. Patients should be followed up by a single physician with experience so that these low-prevalence diseases do not go undetected. If AS is suspected, a hearing test must be performed to rule out bilateral hearing loss for high frequencies (2000–8000Hz).8 When we reviewed the hearing tests, we found that hearing loss was only diagnosed in half of the cases, as it was considered to be normal when the impairment was mild: this is a confounding factor. Another added problem is the transition from Paediatrics to Nephrology for adults, in such a difficult stage as adolescence, when the patient may come to deny his or her disease and reject chronic treatment.20
There is a series of factors that accelerate a decline in kidney function and they should be managed to slow down its progression.21 The factors for progression of ADPKD are: genetic (the PKD1 mutation has a worse prognosis than the PKD2 mutation), kidney volume, HTN,5 proteinuria, inability to concentrate urine and hyperuricaemia.22 The factors for progression of AS are: type of mutation (truncating mutations are more serious), proteinuria, hearing loss and episodes of macrohaematuria.23 Renin–angiotensin system inhibitors are a first-line treatment, as they manage HTN, decrease proteinuria and delay a decline in kidney function in both ADPKD24 and AS.25
In conclusion, it is necessary to create an HKD practice in which these types of kidney disease are followed up by a single physician to ensure a suitable diagnostic and multidisciplinary approach, management of factors for progression and collaboration between hospitals contributing to diagnostic and therapeutic advances in this constantly evolving field. It is necessary to stress the importance of preparing a complete family tree to classify the pattern of inheritance (AD, AR or XL) and the type of kidney injury (cystic, tubular, glomerular or syndromic), which, unlike genetic diagnosis, is available at all hospitals.
Please cite this article as: Martínez Jiménez V, Ramos Carrasco F, Alcázar Fajardo C, Cabezuelo Romero JB. Utilidad de una consulta de enfermedades renales hereditarias: un enfoque diferente basado en el árbol genealógico. Nefrología. 2016;36:217–221.



