





Membranous nephropathy (MN) is a common cause of nephrotic syndrome after kidney transplantation (KT); however, scarce is known regarding post-KT thrombospondin type-1 domain-containing 7A (THSD7A)-positive MN. Herein, we report on a 72-year-old woman with end-stage kidney disease due to chronic interstitial nephritis (1996). In February 2020, she received a second deceased-donor KT, achieving optimal kidney function but presenting early post-KT proteinuria, reaching up to 1800mg/24h six months after transplantation, controlled with renin–angiotensin–aldosterone system (RAAS) blockade. In July 2021, a kidney allograft biopsy revealed features consistent with MN. Immunohistochemical stains showed diffuse and granular THSD7A and C4d deposition in glomerular capillary walls and negative PLA2R and IgG4 staining. No anti-THSD7A antibodies were detected in the serum. The pre-implantation biopsy showed no MN-associated lesions and negative THSD7A staining. Secondary triggers such as malignancy were discarded. The present report illustrates a THSD7A-positive MN in a KT recipient. Despite lacking native kidney biopsy and early presentation, a recurrent MN seemed unprovable due to documented native kidney disease and a long time span between native kidney disease and MN diagnosis. We, therefore, presumed primary de novo disease. Two years after KT, kidney function remains stable, and the patient has reached complete remission of proteinuria.
La nefropatía membranosa (NM) es una causa común del síndrome nefrótico tras el trasplante renal (TR); sin embargo, se conoce poco sobre la NM con trombospondina de tipo 1 que contiene el dominio 7A (THSD7A) positiva en el TR. Presentamos el caso de una mujer de 72 años con enfermedad renal terminal secundaria a nefritis intersticial crónica (1996). En febrero de 2020 recibió un segundo TR de donante fallecido que, tras alcanzar una función renal óptima, presentó una proteinuria precoz, no nefrótica, que llegó a los 1.800mg/24h a los 6 meses del trasplante. En julio de 2021 una biopsia renal reveló características compatibles con NM y la inmunohistoquímica mostró una tinción difusa y granular para THSD7A y C4d en paredes de capilares glomerulares, con PLA2R e IgG4 negativos. Los anticuerpos séricos anti-THSD7A fueron negativos. Se descartaron causas secundarias, como neoplasias. Este caso ilustra una NM THSD7A-positiva en un receptor de TR. A pesar de carecer de una biopsia de riñón nativo y de la presentación temprana, la reproducción de una NM parece improbable debido la enfermedad renal primaria filiada y el largo lapso de tiempo entre el diagnóstico de la enfermedad primaria y el diagnóstico de la NM pos-TR. Por lo tanto, sospechamos una enfermedad de novo. A los 2 años del trasplante, la función renal permanece estable y la paciente ha alcanzado remisión completa de la proteinuria.
Membranous nephropathy (MN) is an antibody-mediated glomerular disease histologically characterized by subepithelial granular immune-complex deposits containing immunoglobulins (mainly IgG), antigens, and complement components. This glomerular injury leads to glomerular filtration barrier disruption with the subsequent development of massive proteinuria.1 Despite 20% of cases being attributed to secondary causes, most are considered primary.1 Approximately a third of the patients with primary MN will progress to end-stage kidney disease (ESKD) and may be suitable for kidney transplantation (KT). A significant breakthrough has been made in understanding its autoimmune nature by identifying several target antigens during the last decade. First, M-type phospholipase A2 receptor (PLA2R)2 and thrombospondin type 1 domain-containing 7A (THSD7A)3 were identified as the deposited antigens, accounting for 70–80% and 2–5% of primary cases, respectively.1,4 Secondly, laser microdissection and tandem mass spectrometry allowed to recently discovering some additional potential antigens.1,4
After KT, MN is a leading cause of nephrotic syndrome in adults, only preceded by transplant glomerulopathy.5 It can occur as a recurrence of the primary disease (rMN) or de novo after transplantation (dnMN).6–9 Anecdotally, some cases of MN present in the donor prior to transplantation have been observed.10 PLA2R has been widely associated with de novo and recurrent MN in the allograft.7,11,12 However, scarce information is available regarding recurrent or de novo MN caused by other antigens such as THSD7A.13
Case reportWe report on a 72-year-old Caucasic woman with a medical history of hypertension, dyslipidemia, and ESKD due to chronic interstitial nephritis secondary to recurrent urinary tract infections (UTIs). No native kidney biopsies were performed. In 1996, she received her first deceased-donor KT. No induction therapy was administrated due to low immunologic risk. Maintenance immunosuppression included cyclosporine, mycophenolic acid, and prednisone. One year after the KT, prednisone was withdrawn. In 2013 the patient developed de novo HLA class II donor-specific antibodies (HLA-DSA). Kidney function remained stable, but due to progressive proteinuria (1–2g/24h), a renal allograft biopsy was performed in 2015. Histological findings were consistent with C4d-negative chronic active antibody-mediated rejection (ABMR) with severe chronic transplant glomerulopathy. She had progressive graft dysfunction and began hemodialysis in 2019.
In February 2020, she received a second deceased-donor KT. Pre-implantation biopsy with frozen sections showed glomeruli without morphological alterations, 5% global glomerulosclerosis, and mild chronic tubulo-interstitial changes. Percentage panel reactive antibodies prior to transplantation were 23%, with no preformed HLA-DSA. The patient received basiliximab induction and maintenance immunosuppression therapy with tacrolimus, everolimus, and prednisone. Nadir creatinine reached 1.7mg/dL, but she presented early recurrent UTIs and fluctuant proteinuria, being difficult to assess in this context, but reaching an average of 1800mg/24h at six months after KT. The physical exam was unremarkable. After initiation of renin–angiotensin–aldosterone system (RAAS) blockade in November 2020, proteinuria dropped to 200–300mg/g. No changes in maintenance immunosuppression therapy were made (Fig. 1).
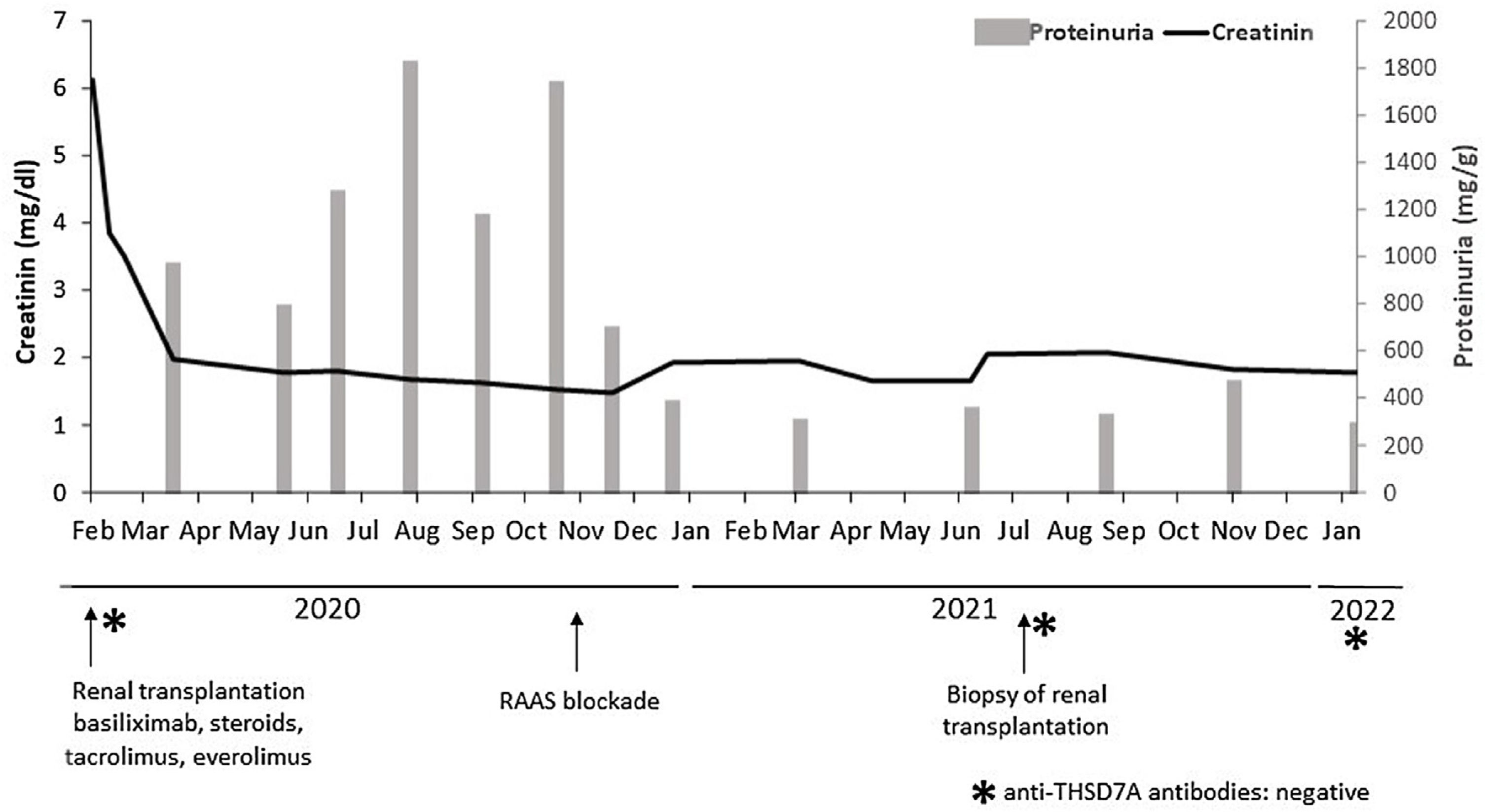
In July 2021, with stable graft function and controlled proteinuria, a one-year protocol allograft biopsy was performed. Light microscopy revealed up to 40 glomeruli, seven with global sclerosis, predominantly in the subcapsular area. Non-sclerosed glomeruli presented normal cellularity and diffuse thickening of peripheral capillary walls. Small spiculated projections (“spikes”) of the membrane were observed with silver stains, and small subepithelial fucsinophilic deposits were detected with Masson's trichrome stains (Fig. 2A–C). On the other side, no double contours of glomerular basement membranes nor mesangial alterations were identified. There was minimal interstitial fibrosis and tubular atrophy and mild acute tubular injury. No microvascular inflammation was observed, and C4d was negative in peritubular capillaries. The immunofluorescence study was unavailable due to the lack of glomeruli in the −80°C frozen sample. Immunohistochemistry in paraffin-embedded tissue showed diffuse and granular THSD7A and C4d staining in glomerular capillary walls (Fig. 2D). PLA2R and IgG4 immunostains were negative in the glomeruli. Serum anti-THSD7A antibodies were also negative. Electron microscopy showed small subepithelial electron-dense immune-like deposits with intervening glomerular basement membrane spikes. Occasionally, the electron-dense deposits where completely surrounded by projections of the glomerular basement membrane (Ehrenreich-Churg stage 2–3). No mesangial, subendothelial nor tubular basement membrane electron-dense deposits were detected.
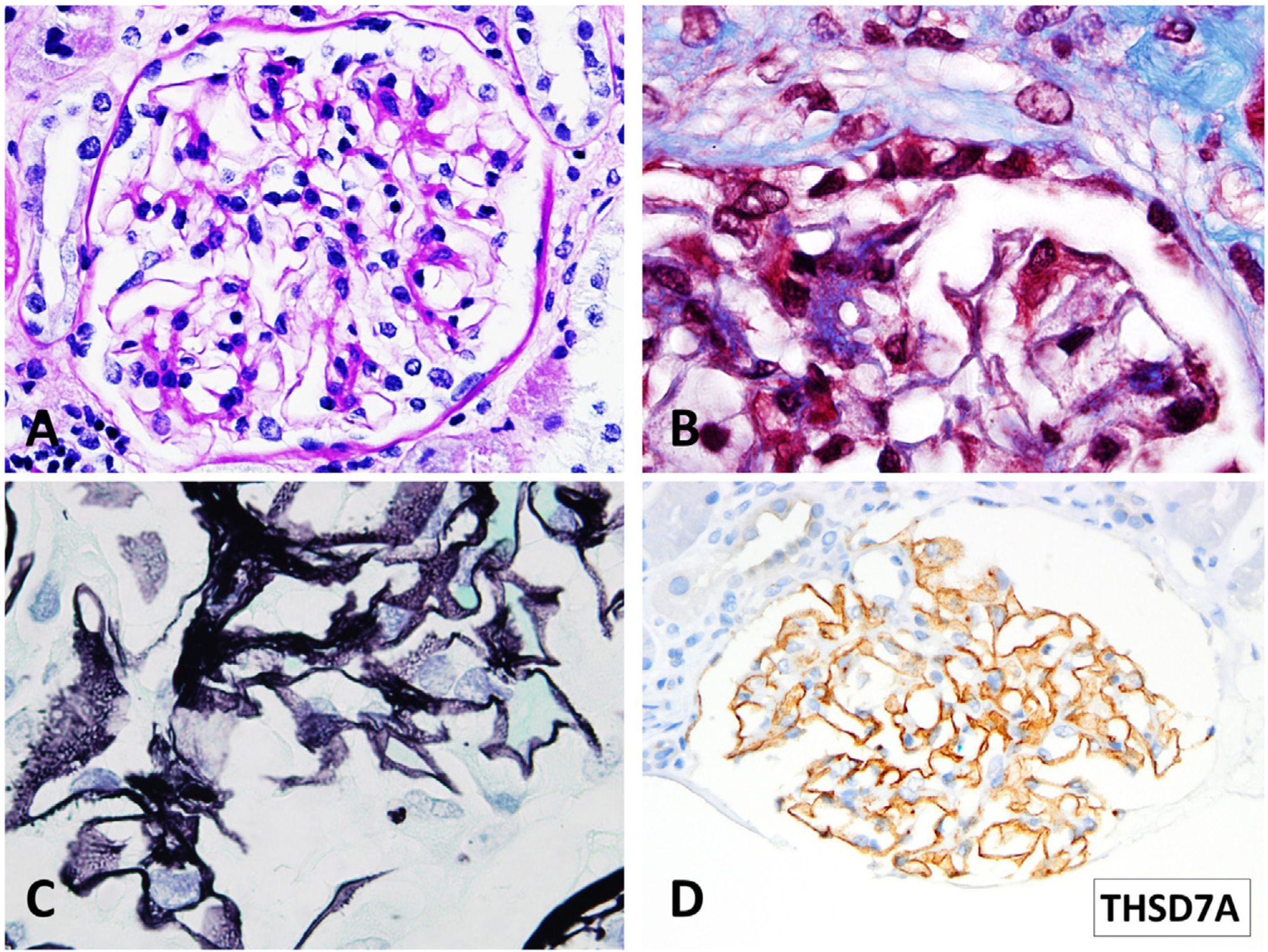
Light microscopy findings. (A) PAS. 40×. Minimal segmental increase in mesangial matrix. Peripheral glomerular capillaries appear stiff and slightly thickened (B) Masson trichrome stain. 100×. Detail of fuchsinophylic deposits in peripheral glomerular capillaries. (C) Silver–Methenamine stain. 100×. Detail of the vacuolated appearance of the glomerular basement membrane. Some small projections (spikes) of silver-positive material in the subepithelial aspect of the glomerular basement membrane are seen. (D) TSHD7A Paraffin immunohistochemistry. 40×. Positive granular stain in peripheral glomerular capillary walls.
The patient was diagnosed with THSD7A-positive MN. Consequently, the pre-implantation biopsy was re-evaluated, showing no MN suggestive lesions and immunohistochemistry in paraffin-embedded tissue showed no THSD7A staining. Likewise, serum anti-THSD7A antibodies at KT tested retrospectively were negative. Infectious, autoimmune, or medication triggers were discarded. Finally, the screening for malignancies (including thoracoabdominal CT, lower gastrointestinal endoscopy, and mammography) showed no additional findings. Despite no native kidney biopsies available and the early presentation after KT, given the medical records regarding the patient's primary kidney disease, the long-time span from primary disease to post-KT MN diagnosis, and the non-nephrotic range proteinuria presentation, we classified the MN as a de novo disease.
DiscussionThis report illustrates a rare case of THSD7A-positive MN in a KT recipient diagnosed by protocol biopsy. Non-nephrotic proteinuria remitted after RAAS blockade initiation before histologic disease confirmation, and no further immunosuppression treatment was needed. To the best of our knowledge, only one previous study has described the presence of THSD7A-positive MN in a kidney allograft.13
THSD7A is a podocyte-specific multidomain transmembrane protein that targets autoimmunity in patients with MN.3 First identified in 2014, THSD7A-positive MN is responsible for about 2–5% of primary cases in native kidneys.1,3,4 The detection of anti-THSD7A circulating autoantibodies has evolved from semi-quantitative tests such as Western Blot and indirect immunofluorescence tests to more sensitive and rapid assays, such as enzyme-linked immunosorbent assay, allowing a quantitative assessment for both diagnosis and follow up of these patients.14,15 Additionally, few specialized laboratories perform biopsy immunohistochemical staining to detect THSD7A expression in the glomeruli.4,15 Positive THSD7A staining is highly sensitive and specific and correlates with serum antibody detection.16
Other antigens have been depicted as a cause of MN after KT,7,17,18 some showing a positive predictive value for recurrence when detected pre-KT.11,12 Moreover, in a recently reported case of post-KT NELL1-positive MN, the authors outlined a serum correlation between the antigen and urinary protein excretion and suggested it might be of predictive value preceding complete remission.17 Little is known about anti-THSD7A antibodies and the risk of recurrence or dnMN after KT. Tomas et al. reported a 57-year-old male with early nephrotic-range proteinuria due to recurrent THSD7A-positive MN. The patient had detectable anti-THSD7A antibodies at KT that remained detectable during the follow-up and despite immunosuppressive treatment.13 Whether increased pre-transplant anti-TSHD7A titles may correlate with post-KT recurrence is unknown. Still, in their study, Tomas et al. demonstrated that human anti-THSD7A antibodies could induce an MN-histopathological pattern in mice, causing foot process effacement and consequent proteinuria.13 In the present case, serum anti-THSD7A antibodies were undetectable at transplantation and KT biopsy. Nevertheless, whether this could be associated with a better outcome is unknown. There is a lack of evidence regarding the discrepancy between positive staining for THSD7A in the graft biopsy and negative circulating anti-THSD7A. However, this discordance has previously been described in PLA2A-associated MN. Interestingly, some authors have described that patients with persistent disease activity presented seroconversion of the antiPLA2R antibodies during follow-up.19,20 The authors attribute this circumstance to the kidney as a sink hypothesis: the serum antibodies only become positive when the kidney buffering capacity has been saturated.19 Furthermore, serum anti-THSD7A negativity at the time of KT biopsy could result from complete remission of proteinuria after treatment,1 similar to the NELL1-positive MN case.17 Unfortunately, anti-THSD7A antibodies were not tested concomitantly with the proteinuria peak, but we cannot assure that MN caused the sub-nephrotic proteinuria due to other cofounders such as UTIs and everolimus.
In the absence of a native kidney biopsy, the differential diagnosis between dnMN, recurrence of an undiagnosed primary disease, or MN present in the donor may be challenging. Unfortunately, in Spain a large proportion of patients do not have a renal biopsy available as, in a population with an annual incidence of renal replacement therapy initiation of 126 patients per million population21 the annual rate of renal biopsy in 1994–1999 was 4.8 per 100,000 population-years.22 Re-evaluation of the pre-implantation biopsy in our patient showed no morphological MN lesions and negative THSD7A immunostaining. Besides, retrospectively analysis of anti-THSD7A antibodies at KT, which would have favored a recurrent disease, was negative. Disregarding the early presentation after the second KT, the possibility of a rMN seemed unlikely due to the patient's medical records concerning primary kidney disease, the long span between primary disease and the MN diagnosis (25 years), and the non-nephrotic range proteinuria at presentation. We, therefore, suspected de novo disease. De novo MN in kidney allografts has been associated with ABMR due to excessive antigen exposure and subsequent antigen-antibody formation. Nonetheless, the relationship between rejection's pathogenic role and MN-specific antigens stays undefined.8 Of note, our patient had no other ABMR-related histological lesions nor HLA-DSA.
Comparable to NELL1 antigen, THSD7A-positive MN has indeed been associated with malignancies in 20% of cases.14 THSD7A overexpressed in some malignancies may trigger the immune response that yields MN in the kidney.1 In the present case, screening for malignancies showed no abnormalities, and no other systemic conditions were detected.
Therapeutically, the patient started RAAS blockade due to non-nephrotic proteinuria. Although early initiation of Rituximab is strongly recommended in patients with rMN, especially if proteinuria increases over 1g,5,9 the best therapeutic approach in patients with dnMN is still unclear. Herein the patient had an excellent response to RAAS blockade intake resulting in complete remission of the proteinuria when the biopsy was performed. Up to date, no other treatments have been needed, and the immunosuppressive regimen has remained unchanged.
In conclusion, the present report illustrates an unusual case of THSD7A-positive MN in a KT recipient. Given the nonexistent triggers and the unlikeliness of recurrence, we presume this case illustrates a primary de novo disease. Two years after KT, the patient maintains stable kidney function and has reached complete remission after RAAS blockade. Serum anti-THSD7A antibodies remain negative 6 months after diagnose (Fig. 1) and no additional biopsies have been performed so far. However, the prognosis of this entity occurring after transplantation is still unknown.
Data availability statementThe authors confirm that the data supporting the findings of this case report are available upon request.
FundingAB has support from a Rio Hortega contract (CM19/00004, ISCIII), a M-AES grant (MV20/00072, ISCIII), and a Spanish Society of Nephrology scholarship. The present study has been supported by funding from projects PI20/00090 (Spanish Ministry of Health ISCIII FIS-FEDER) and RD16/0009/0013 (ISCIII FEDER REDinREN). MC has support from a grant Spanish Ministry of Health ISCIII FIS-FEDER number INT21/00003.
Conflicts of interestThe authors declare no financial disclosures or conflicts of interest.
We thank the patient for consenting and sharing her clinical course and medical data. We are indebted to Rosa Causadías, Anna Bach, Guillermo Pedreira, Aida Martínez, Anna Faura, and Marisa Mir for their assistance with patients.



