

Parvovirus B19 (PB19) is a single-stranded DNA virus for which 90% of the population has antibodies, given that primary infection usually occurs during childhood. While PB19 infection is usually trivial in the general population,1 in immunosuppressed patients it may manifest itself in the form of pure red cell aplasia or pancytopenia (due to the trophism of the virus towards the P antigen on the surface of erythroblasts).2–4 It is also a cause of thrombotic microangiopathy (TMA) regardless of the degree of immunosuppression. We present here a case of pancytopenia secondary to PB19 complicated by TMA in a kidney transplant patient after withdrawal of immunosuppression.
This was a 48-year-old male with a history of chronic kidney disease resulting from chronic interstitial nephropathy in the context of repeated urinary tract infections and urinary tract malformations, having had his first and only kidney transplant and undergone a urostomy in 2014, on immunosuppression with prednisone, tacrolimus and mycophenolic acid.
Over the course of follow-up, he had progressive deterioration in renal function to baseline creatinine levels of around 4 mg/dl in association with chronic antibody-mediated rejection in July 2020 and repeated urinary tract infections related to the malformations described above.
The patient was admitted in October 2020 for asthenia and fever. Lab tests revealed a deterioration in renal function with a creatinine level of 8.36 mg/dl, metabolic acidosis, haemoglobin of 3.8 g/dl, 98,000 platelets/μl and 3200 leucocytes/μl. With these findings, he was given three units of packed red blood cells and restarted on chronic haemodialysis via a central venous catheter.
Subsequent investigation of the anaemia revealed microcytic and hyporegenerative anaemia, with no red blood cell schistocytes in the smear. However, suppressed haptoglobin and an LDH of 511 U/l were found. In view of these data, an endothelial cell culture was performed, but was negative. Coombs test, serum electrophoresis, Leishmania and PB19 serologies and viral loads for cytomegalovirus, BK virus and Epstein-Barr virus were negative.
With no improvement ten days after admission, a bone marrow biopsy was performed, showing hypocellular marrow with erythroid hyperplasia, lantern cells and viral inclusions positive for anti-PB19 antibodies (Fig. 1). Tacrolimus and mycophenolic acid were discontinued and intravenous immunoglobulins (IVIg) were started at a dose of 400 mg/kg/day for five days.
 (haematoxylin–eosin stain). The image on the right shows parvovirus B19 viral inclusions (arrowheads).")
After completing the treatment, the patient's anaemia and thrombocytopenia worsened and he developed difficult-to-control hypertension (HTN), with an increase in LDH to 655 mg/dl. In addition, 6.3% reticulocytes (consistent with hyperregenerative anaemia) and three red blood cell schistocytes per field were observed in the peripheral blood smear. Donor-specific antibody testing was negative and other causes of HTN were ruled out by renal doppler.
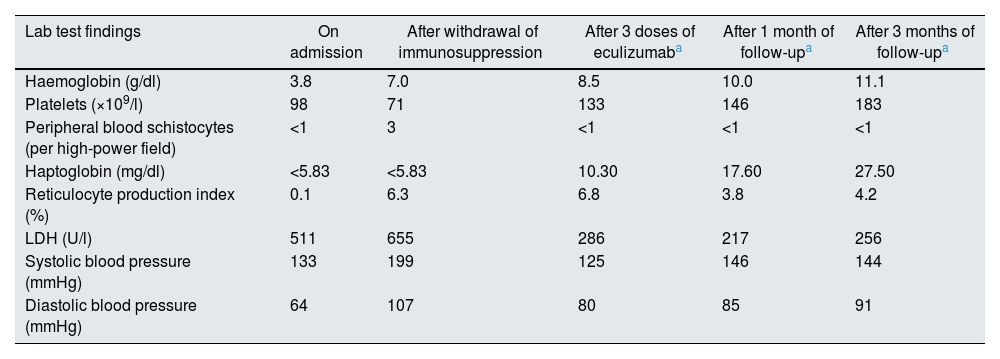
Given the temporal relationship with the withdrawal of immunosuppression and the impossibility of performing a renal biopsy due to the high risk of bleeding, the patient was given 900 mg of eculizumab empirically every week for three weeks, achieving resolution of the haematological parameters and arterial hypertension (Table 1). However, no recovery of renal function was observed, probably because the patient already had advanced chronic kidney disease. Therefore, at discharge, the patient remained dependent on chronic haemodialysis.
Changes in the patient’s clinical and biochemical parameters on admission, after withdrawal of immunosuppression, after three doses of eculizumab and during subsequent follow-up.
| Lab test findings | On admission | After withdrawal of immunosuppression | After 3 doses of eculizumaba | After 1 month of follow-upa | After 3 months of follow-upa |
|---|---|---|---|---|---|
| Haemoglobin (g/dl) | 3.8 | 7.0 | 8.5 | 10.0 | 11.1 |
| Platelets (×109/l) | 98 | 71 | 133 | 146 | 183 |
| Peripheral blood schistocytes (per high-power field) | <1 | 3 | <1 | <1 | <1 |
| Haptoglobin (mg/dl) | <5.83 | <5.83 | 10.30 | 17.60 | 27.50 |
| Reticulocyte production index (%) | 0.1 | 6.3 | 6.8 | 3.8 | 4.2 |
| LDH (U/l) | 511 | 655 | 286 | 217 | 256 |
| Systolic blood pressure (mmHg) | 133 | 199 | 125 | 146 | 144 |
| Diastolic blood pressure (mmHg) | 64 | 107 | 80 | 85 | 91 |
LDH: lactate dehydrogenase.
Discharge serology for PB19 was positive and subsequent testing for mutations of alternative complement pathway regulatory proteins was negative.
PB19 infection usually occurs in the first year after transplantation2 and should be suspected in patients with erythropoietin-resistant anaemia.2,4,5 For diagnosis, the guidelines recommend determining viral load, as serology is not very sensitive in immunosuppressed patients.2 In this case, a bone marrow biopsy was necessary given the low initial suspicion for this infection due to the amount of time since transplantation. The most common findings in bone marrow biopsy are giant proerythroblasts with intranuclear inclusions (lantern cells) (Fig. 1).
The recommended treatment2 is IVIG at a dose of 0.4 g/kg/day for five days. In most cases, this treatment allows patients to be transfusion-independent at 12 months.6
After the start of treatment, the patient developed TMA. PB19-associated TMA is most commonly reported in the immediate post-transplant period.7 Two mechanisms have been proposed as possible explanations for the development of PB19-associated TMA: direct invasion of the endothelium via the P antigen7,8 or immune complex formation. In both cases, endothelial damage would stimulate activation of the alternative complement pathway, which would explain the good response to a short course of eculizumab.9
In conclusion, PB19 can trigger bone marrow aplasia in the late post-transplant period and is a rare cause of TMA which, in this case, showed significant improvement with eculizumab.



