


INTRODUCTION
Signal transduction in different types of cells often involves conditional or constitutive activation of receptor tyrosine kinases, which trigger multiple cytoplasmic kinases. Such signalling pathways can operate independently, in parallel, and/or through interconnections that promote different diseases to develop. The most important signalling pathways are phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC) and mitogen-activated protein kinase (MAPK)/Ras.1
mTOR PROTEIN
mTOR (Mammalian Target of Rapamycin) is a 289-kDa serine/threonine protein kinase. The TOR family of proteins has pleiotropic functions, and participates in the regulation of the initiation of mRNA transcription and protein translation in response to intracellular concentrations of amino acids (AA) and other essential nutrients. It is involved in the organisation of actin cytoskeleton, membrane trafficking, protein degradation, PKC signalling and ribosome biogenesis. mTOR regulates essential signalling pathways and is involved in coupling growth stimuli and cell-cycle progression.
There are two complexes that contain mTOR: a rapamycin-sensitive complex (mTORC1), which is defined by its interaction with the protein raptor (regulatory-associated protein of mTOR), and a rapamycin-insensitive complex (mTORC2), defined by its interaction with rictor (rapamycin-insensitive companion of mTOR). mTOR is a key kinase which acts downstream of the activation of PI3K. Much evidence supports the hypothesis that mTOR is the key for cellular catabolism and anabolism, determining whether cells, and in particular carcinogenic cells, grow and proliferate. Furthermore, mTOR regulates apoptosis.
mTOR, in the shape of the two signalling complexes, cited as mTORC1 and mTORC2 with their two different proteins, raptor and rictor, establish two different mTOR pathways. The raptor-mTOR pathway (mTORC1) regulates cellular growth (cellular mass accumulation) and proliferation through P70S6K and 4E-BP. It responds to nutrients and growth factors, partly due to regulators like TSC1/TSC2 (tuberous sclerosis complex 1: hamartin; tuberous sclerosis complex 2: tuberin) and Rheb (a Ras family GTPase). mTOR (mTORC1) is phosphorylated by AKT (also called protein kinase B-PKB) through inactivation of the tuberous sclerosis complex (TSC) and is directly activated by Rheb.2,3
The rictor-mTOR (mTORC2) complex regulates AKT/PKB, PKCα, Rho/rac, to control cell polarity and the cytoskeleton. Growth factors and AA activate AKT and mTOR through PI3K.1
There are transcription factors that could be activated or inhibited through AKT phosphorylation. AKT activates the NF-kB transcription factor, which increases the transcription of antiapoptotic genes. The NF-kB transcription factor is the central mediator of the immune response, inflammatory response and cell survival response. Following its activation, IKK phosphorylates IkB, resulting in their ubiquitination and degradation in the proteasome. This exposes the NF-kB nuclear localisation sites and allows it to translocate to the nucleus to induce antiapoptotic gene expression. Growth factors, such as the vascular endothelial growth factor (VEGF), activate NF-kB and protect against apoptosis. On the other hand, NF-kB inhibition sensitises the cell to a wide variety of proapoptotic stimuli.3
ROLE OF mTOR IN ACUTE RENAL FAILURE
Regeneration and restoration of the morphology and renal function partly depends on the capacity of the remaining viable kidney tubule cells to proliferate and restore the damaged epithelium.4 mTOR is an ubiquitous kinase and its inhibition by rapamycin also blocks proliferation, including cells in the kidney.5 mTOR plays an important role in the regeneration and repair process following an experimental acute kidney injury. The mTOR activity is low or absent in the normal kidney, but increases significantly following an ischaemia-reperfusion process. Inhibition of mTOR by rapamycin delays renal recovery-repair.5
ROLE OF mTOR IN CHRONIC KIDNEY DISEASE
The mTOR pathway plays an important role in the mechanisms that are involved in chronic kidney disease (CKD) progression caused by diabetes, for instance. Rapamycin reduces interstitial inflammation, fibrosis and loss of kidney function associated with CKD progression.
Several studies have shown why it is important for mTOR to be activated in physiological and pathological forms of renal hypertrophy and other organs, including hypertrophy of the diabetic nephropathy (DN).6 This phenomenon contributes to podocyte damage and progressive loss of renal function.7 Furthermore, upon mTOR activation, an increase in matrix protein synthesis will contribute to glomerular basement membrane thickening and the accumulation of the mesangial matrix, which are characteristic of DN.8 mTOR activation in diabetes is, at least, partly caused by hyperglycaemia via AKT activation.8 Rapamycin has not only reduced mTOR activity in in vivo models, but it has also reduced the characteristic DN changes mentioned above, and it is associated with a reduction in albuminuria.9
Similar phenomena can be observed in non-diabetic CKD, with an increase in the proinflammatory and profibrotic cytokine expression, interstitial inflammatory cell infiltration and renal fibrosis.10 Rapamycin treatment for membranous glomerulonephritis in rat models reduces all of these phenomena.11
ROLE OF mTOR IN AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disease characterised by the formation of multiple cysts within the renal parenchyma, which results in renal failure. It affects up to one case in every 400-1000 newborns. ADPKD is related to PKD1 and PKD2 gene mutations, which are located in chromosomes 16 and 4, respectively. The PKD1 gene codes for the transmembrane protein polycystin-1 (PC1). It has been reported that this protein is involved in cell-cell adhesion, cell-matrix adhesion, transduction of intracellular signals and polycystin-2 regulation (PC2), as the PKD2 gene codes for the PC2 protein, which is a calcium channel.2,12 The PC1 and PC2 complex is essential for maintaining the physiological phenotype of the tubular epithelial cells.
The polycystin protein complexes are fundamental for maintaining intracellular calcium homeostasis. All renal tubular epithelial cells (except the interspersed ones) were recently found to have a single primary cilium which has mechanoreceptor and chemoreceptor functions. Stimulation of these cilia increases the intracellular calcium through the PC1 and PC2 complex. Intracellular calcium controls many of the cellular processes, such as proliferation. Therefore, drugs that reduce cyclic adenosine monophosphate (cAMP) or which increase intracellular calcium could treat ADPKD. Calcium is also involved in signalling pathways related with growth factors, activating signalling cascades for some protein kinases. Alongside this, PC1 regulates the mTOR activity (loss of PC1 activity in ADPKD allows significant mTOR activity inside the cyst epithelial cells in mouse and human models), which would be a therapeutic target.2,13
RAPAMYCIN DEVELOPMENT
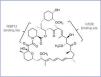
Rapamycin is also called sirolimus; it is a natural antibiotic synthesised by S. hygroscopicus. This bacterium was discovered 30 years ago in earth from Easter Island, Rapa Nui, which is where the name rapamycin came from. It is a lactone initially developed as an antifungal agent. In its purest form, it resembles a white crystalline powder, insoluble in an aqueous solution, but soluble in organic solvents. The chemical structure is shown in Figure 1.
Between 1982 and 1988, rapamycin was developed as an immunosuppressive agent. Thanks to these studies, the mechanism of action of this molecule was clarified. Rapamycin interacts with the immunophilin FK506 binding protein (FKBP12) through its methoxy group. The rapamycin-FKBP12 complex is specifically bound to the mTOR protein, inhibiting the effector signalling pathways dependent on said protein.14 Rapamycin inhibits antigen-induced T cell proliferation and the cytokine-induced proliferative responses, including interleukin-16 (IL-16), immunoglobulin growth factor (IGF), etc. It follows the cytochrome CYP450 3A4 pathway as the main system responsible for drug biotransformation, generating inactive metabolites.
A high level of synergism has been shown for this drug with cyclosporin,15 both in vivo and in vitro. As such, the dosage for effective immunosuppression is reduced, decreasing the probability of kidney graft rejection and minimising cyclosporine-induced toxicity.16
An important aspect of rapamycin as an immunosuppressant is that it does not produce secondary effects on renal haemodynamics. Treatment with rapamycin maintains glomerular filtration and the renal blood flow both for normal rats and rats with salt depletion or spontaneous hypertension.17 The renal tissue seems to be protected during rapamycin treatment by an inhibition of the intrarenal angiotensin II cascade. However, rapamycin does produce a dose-dependent tubular toxicity in rats (including hypercalcaemia and hypophosphataemia), a phenomenon linked to delayed recovery of the tubular epithelial function after injury.18
Rapamycin was approved by the US Food and Drug Administration (FDA) in 1999 as a preventative treatment of acute rejection in combination with cyclosporine and steroids. A year later, the drug was approved by the European Medicines Agency (EMEA) as an alternative to calcineurin antagonists in long-term treatment to prevent graft rejection. Rapamycin, unlike cyclosporine, does not seem to increase the risk of malignancy, but reduces the risk of lymphoproliferative processes after transplantation (reducing AKT levels). However, rapamycin increases CsA side effects: high blood pressure, acne and hirsutism and has been associated with mild secondary effects such as diarrhoea, tachycardia, oedemas, dyslipidaemia and non-infectious pneumonitis.
In addition to its immunosuppressant capability, rapamycin has been proven to act as a preventative agent on restenosis of the coronary arteries.19 The hypothetical mechanism responsible for the inhibition of vascular smooth muscle cell proliferation by rapamycin includes, among others, the binding to protein FKBP12.
In another article published in this issue, Dr Cabrera et al20 discuss the results of rapamycin treatment on the evolution of angiomyolipomas in a substantial number of patients suffering from tuberous sclerosis (TS) or Pringle-Bourneville disease (this number is considerable taking into account the prevalence of the disease).
The TS complex is a systemic disease, which is an autosomal dominant genetic disorder with a prevalence of one case for every 6000 live births.21 It is characterised by benign tumours (hamartomas) in multiple organs and systems, including brain, skin, kidney, lungs, heart and retina. Angiomyolipomas are tumours rich in adipose tissue, muscle and blood vessels that may bleed or infiltrate the kidneys causing deterioration of kidney function for up to 80% of patients.22
Mutations that may occur in any of the two TS genes, TSC1 (hamartin) or TSC2 (tuberin) is above 85% for TS patients.23 Proteins coded by these two genes form a tumour-suppressive complex, acting through Ras homolog enriched brain protein (Rheb), which limits mTOR activation (mTORC1). When TSC1 or TSC2 are deficient, mTORC1 is overexpressed constitutively, provoking cellular growth, proliferation and abnormally high protein synthesis.24
The clinical trial in phase 4 which the above mentioned authors present,20 shows a significant reduction in the volume of angiomyolipomas after as little as six months of rapamycin treatment (55.1% average), reaching a reduction of 66.3% at 12 months. The reduction of volume of angiomyolipomas seems to be due to the effect of mTOR inhibition and its effect on VEGF. Another interesting point that these authors document in this paper is the possibility of reducing the rapamycin dose once the peak reduction of the angiomyolipomas volume has been achieved, seemingly being at between 12 and 24 months.20,25 However, the treatment must not be withdrawn as that would promote tumour regrowth.25,26 Whether the effect of rapamycin would be the same in any type of angiomyolipoma is still unknown, depending on its size and location (unilateral or bilateral). Furthermore, as there are no genetic studies, we are still unaware if the rapamycin response varies depending on whether it is located in TSC1 or TSC2, given that the phenotype may be different.
Only one patient was excluded before 12 months due to reactivation of an erythema nodosum. No changes were found in renal function, as rapamycin plasma levels were maintained constant. Despite the adverse effects cited in the medical literature,25 the authors found a higher incidence of oral aphtae and dyslipidemia.20 Furthermore, facial angiofibromas in these patients are smaller and not as relevant.
There are very few data in the literature on this matter, and those found are isolated and based on some clinical cases26 and a trial published in the New England Journal of Medicine with 25 patients, of which only 20 completed the 12-month follow-up period.25 None of these references presented genetic studies. Nevertheless, as the authors cited in their article,20 there are several studies being developed in the United States and Europe.
Rapamycin, through mTOR inhibition, is an alternative therapy for this disease. More studies are needed to define the risks and benefits of long-term rapamycin treatment for this type of genetic disorder, its use as a monotherapy or in combination, and considering location and size of the angiomyolipomas.
KEY CONCEPTS
1. mTOR is an important modulator of several types of kidney diseases.
2. mTOR is activated following acute kidney damage and contributes to renal regeneration and repair. Rapamycin can delay renal recovery and repair.
3. mTOR plays an important role in the formation and growth of cysts in ADPKD.
4. Rapamycin may, through different mechanisms, delay the reduction of glomerular filtration in chronic kidney diseases, reducing their progression.
5. When used as a monotherapy, rapamycin can be an alternative therapy for preventing the growth of kidney angiomyolipomas in tuberous sclerosis and delay/prevent renal failure. Possible adverse effects of rapamycin must be taken into consideration.
6. Although intervention on the mTOR complex in progression of chronic kidney diseases may seem straightforward, more studies must be performed to establish this interconnection.

Figure 1. Chemical structure of rapamycin



