


En la actualidad se reconoce que contaminantes ambientales como el cadmio, el plomo y el arsénico tienen un papel importante en la génesis de la insuficiencia renal crónica. Estudios epidemiológicos han demostrado la fuerte asociación entre exposición a estos metales y la presencia de daño renal crónico. Los mecanismos fisiopatológicos de daño renal por metales son complejos y aún se desconocen varios aspectos de su metabolismo y mecanismos de daño en el organismo. Es objetivo de esta revisión analizar dichos mecanismos fisiopatológicos de daño renal por cadmio, plomo y arsénico.
We currently recognise that environmental toxins such as cadmium, lead, and arsenic play a significant role in the development of chronic renal failure. Epidemiological studies have shown a strong association between exposure to these metals and the presence of chronic kidney injury. The physiopathological mechanisms behind metal-induced kidney injury are complex, and some aspects of their metabolism and damage mechanisms remain unknown. This review aims to analyse the physiopathological mechanisms of kidney injury due to cadmium, lead and arsenic.
INTRODUCTION
Both the incidence and prevalence of chronic renal failure have risen constantly over the last 3 decades, which is now a growing public health problem. Identifying risk factors associated with the disease is essential in order to prevent it from affecting even more patients.
Studying the toxic effects of heavy metals on the human body has become especially important in the last 50 years, given that large amounts of these products were disposed of as industrial waste and they are not biodegradable, remaining in the environment for long periods of time. For this reason, despite the fact that strict regulations enforced mainly in Europe and North America limit the disposal of heavy metals, high levels of these elements are still present in soil and sediment, resulting in chronic exposure in the general population.
Heavy metals are a poorly defined group of elements. Some are necessary for the human body, such as iron (Fe), cobalt (Co), copper (Cu), manganese (Mn), molybdenum (Mb) and zinc (Zn). It is unknown whether the other metals – lead (Pb), cadmium (Cd) and arsenic (As) – serve any purpose in the body, but they do have a direct effect on the kidneys and they are particularly nephrotoxic, even at “normal” levels. There is no clear evidence of nephrotoxicity due to other metals such as uranium and mercury.1
The aim of this review is to analyse the epidemiology, physiopathology and clinical manifestations of nephrotoxicity associated with these metals.
ABSORPTION AND METABOLISM OF DIVALENT METALS
Intestinal absorption of divalent metals such as Cd and Pb is facilitated by divalent metal transporter 1 (DMT-1). DMT-1 is located in the duodenum, erythrocytes, liver and cells in the proximal convoluted tubule (PCT). This protein transports Fe and has a high affinity for other divalent metals such as Cd, Ni (nickel), Pb, Co, Mn, Zn and Cu.2 Decreased intake of Fe and Zn results in increased expression of DMT-1, which increases intestinal absorption of Cd and Pb and therefore toxicity by these metals.3 Experiments in cell lines in which DMT-1 expression has been blocked suggest that there is a different Pb transporter.4
Heavy metals are metabolised in the liver, where they bind to low molecular weight proteins (<10kDa) called metallothioneins (MT). These proteins are widely distributed throughout the body and contain a large quantity of the amino acid cysteine, which gives them a high affinity for reacting with and storing metals such as Zn, Cd, Hg (mercury), Cu, Pb, Ni, Co and Fe.
The main function of MT is to store essential metals such as Zn and Cu in the intracellular medium and transfer them to metalloproteins, transcription factors and enzymes. MT also play a role in the elimination of free radicals and in cellular repair and regeneration processes.5 Increased intracellular levels of Cd and Pb increases MT expression, and MT knockout mice are more susceptible to toxicity from these metals.6
CADMIUM NEPHROTOXICITY
Cd is one of the most toxic elements to which humans are exposed. Environmental exposure mainly occurs by contact with tobacco smoke, water and foodstuffs such as vegetables, grains and molluscs. This metal gradually accumulates in the body and levels increase with age given its long half-life, which is more than 20 years.7
Epidemiology
Various epidemiological studies have demonstrated that environmental exposure to Cd increases the risk of developing kidney injury. During the 1950s in Japan, doctors began to recognise an association between environmental exposure to Cd and increased numbers of women with kidney disease characterised by tubular dysfunction, chronic kidney disease, and a type of osteomalacia known as “itai-itai”.8 It was later found that workers suffering from industrial exposure to Cd had an increased risk of developing kidney disease.9 However, it was not until the publication of Bernard’s studies in Belgium that it was understood that exposure to even low Cd levels had nephrotoxic effects and that up to 7% of the exposed population suffered kidney injury.10
Järup et al11 studied 1021 people and demonstrated that the prevalence of tubular damage marker alpha 1-microglobulin was significantly higher in subjects whose urinary excretion of Cd was within the high range of normal limits (odds ratio [OR]: 6, 95% confidence interval [CI]: 1.6-22). Noonan et al12 showed the same relationship between normal-to-high urinary levels of Cd and the presence of NAG (N-acetyl-beta-D-glucosaminidase) and alanine aminopeptidase tubular dysfunction markers. The literature does not currently include reports on the effects of Cd on the progression of chronic kidney injury, although a Swedish study by Hellstrom et al13 showed a higher incidence rate of patients on dialysis (OR: 18, 95% CI: 1.3-2.3) among people exposed to Cd than among those with no Cd exposure.
In addition to its nephrotoxic effect, Cd is also associated with increased risk of developing diabetes,14 cancer, and cardiovascular disease. In a sample of 13 958 adult participants in NHANES III (National Health and Nutrition Examination Survey III), Menke et al15 showed that high levels of Cd in urine were associated with higher overall mortality and increased risk of cancer. Exposure to Cd also increases the risk of high blood pressure and it is considered a risk factor for cardiovascular mortality and morbidity.16
Physiopathology
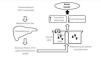
Cd in food is bound to metallothionein and phytochelatin proteins, which are involved in vacuole confinement of heavy metals in vegetables. These proteins are broken down by the action of the gastric juice, releasing Cd that will be absorbed in the intestine by DMT-1 and ZIP-8 transporters.17,18
In circulating blood, it binds to albumin and is transported to the liver, where it binds to glutathione (GSH) and metallothionein-1 (MT-1). The Cd-MT-1 complex is secreted in bile and subsequently reabsorbed into the blood by means of enterohepatic circulation. Cd-MT-1 is a low molecular weight complex (<7kDa) which is easily filtered by the glomerulus and is entirely reabsorbed in the S1 segment of the PCT by endocytosis in a process mediated by the proteins megalin and cubilin.19
The ZIP-8 transporter is also located in PCT cells, and it is able to transport Cd and other divalent metals through the apical membrane of these cells; however, the role it plays in Cd toxicity is unknown.17
Within the intracellular medium of PCT cells, the Cd-MT-1 complex is stored and broken down by lysosomes. Free Cd is then transported to the cytoplasm by lysosomal DMT-1.6 Activation of protein kinase C increases expression of DMT-1, thereby increasing tubular toxicity by Cd.20
Free Cd accumulates in mitochondria, blocking the respiratory chain at complex III. This results in mitochondrial dysfunction and the formation of free radicals, which activates caspase enzymes and the apoptosis process. Free Cd also binds to protein sulfhydryl groups and affects the structure and function of the proteins. It has been demonstrated that Cd interferes with enzymatic activities of the calcium-calmodulin complex, inhibits Na+-K+-ATPase activity, and stimulates activity by MAP kinases. In paracellular tight junctions, it affects the distribution of paracellular tight junction proteins and decreases transepithelial resistance.21,22
Only 10% of filtered Cd is reabsorbed into distal ends of the nephron, and it is possible that the Cd 's hypercalciuric effect is the result of inhibition of calcium channel activity in the distal tubule.23
Another nephrotoxicity mechanism is the one mediated by the formation of anti-MT antibodies; exposure to Cd increases MT production in the liver and kidneys, which constitutes a protective response to limit its toxicity. However, once the MT’s capacity for Cd storage has been exceeded, free Cd is able to induce the formation of antibodies against MT, which are also toxic to PCT cells (Figure 1).24
The effect of foetal exposure to Cd is unknown. Jacquillet et al25 showed that an offspring of rats exposed to Cd during gestation had decreased renal function, proximal tubular damage and abnormal paracellular tight junctions in the glomeruli in adulthood, as well as PCT characterised by alterations in the expression and arrangement of claudin-2 and claudin-5.
Clinical manifestations
The main effects of chronic Cd toxicity are kidney injury, bone demineralisation, high blood pressure, pulmonary function disorders (mainly obstructive) and different types of cancer (bladder, lung, etc.)
In the kidney, Cd mainly affects PCT cells. This damage manifests clinically as low molecular weight proteinuria, aminoaciduria, bicarbonaturia, glycosuria and phosphaturia. Tubular damage markers such as alpha-1-microglobulin, beta-2-microglobulin, NAG and KIM-1 (kidney injury molecule-1) are useful in detecting early tubular damage.26
People with incipient renal injury are more susceptible to the nephrotoxic effects of Cd.27 In patients with diabetic nephropathy, urinary excretion of CD is directly related to increased urinary excretion of beta-2-microglobulin and albuminuria.28
Determining Cd levels in the bloodstream is used to diagnose acute exposure, whilst urinary excretion of Cd is used to assess Cd body burden and is useful for evaluating chronic exposure.29
Prevention is the most important factor in the management of exposure to this metal, since there is no effective means of treating Cd toxicity.
LEAD NEPHROTOXICITY
The toxic effects of Pb have been known for more than 2000 years, since lead intake was a common problem among the Romans. At present, exposure to high concentrations of Pb is less common, due to better industrial management and the fact that Pb is no longer added to paint and petrol. However, Pb contamination is still a public health problem in many countries in Africa, Asia and Latin America due to domestic exposure through contaminated water and soil.30
Epidemiology
The first reported case of nephrotoxicity associated with Pb was described in the 19th century. Since then, exposure to high concentrations of Pb has been considered a risk factor for developing high blood pressure and kidney injury. However, it was not until recent times that studies recognised that exposure to “normal” levels had a direct effect on kidney function and increased the risk of cardiovascular morbidity.31
Based on results from the NHANES III study, Menke et al32 monitored a population over 12 years and demonstrated that the higher the Pb levels, the higher the mortality rates (mainly due to cardiovascular problems).
In a population of 4813 patients with high blood pressure, Muntner et al33 found increased risk of chronic renal failure (OR: 2.6, 95% CI: 1.5-4.45) in those with higher serum Pb levels. Follow-up studies carried out in Taiwan by Lin et al34,35 found that individuals with chronic nephritis (glomerular filtration rate [GFR]<60ml/min) and high levels of Pb in the body experienced faster deterioration of renal function, and also that chelation therapy with ethylenediaminetetraacetic acid (EDTA) decreased kidney injury progression.
Establishing the maximum non-toxic levels of Pb in blood and urine remains a matter for debate, since there is increasing evidence suggesting that levels previously considered to be non-toxic are associated with higher morbidity and mortality rates in the general population.31
Physiopathology
Pb is mainly absorbed by the intestine and the respiratory system and, to a lesser extent, through the skin. Intestinal absorption is mediated by DMT-1 and increases with deficient intake of Fe and Zn. The respiratory system is a highly efficient route of absorption, with an uptake rate of more than 40% of inhaled Pb; however, the molecular mechanism by which Pb is absorbed is unknown.
Once in the blood, 99% of Pb binds to proteins in the erythrocytes and it is distributed to soft tissue and bone. Bone is the main reservoir for lead in the body and Pb transport to the bloodstream increases during times with the highest bone turnover, such as adolescence and pregnancy.31 Urinary excretion is the main route of Pb elimination from the body.
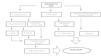
Pb bound to low molecular weight proteins (<1% of the total) is filtered freely at the glomerulus and is reabsorbed by PCT cells by endocytosis. Within the cell, Pb causes mitochondrial damage, formation of free radicals, intracellular depletion of GSH and apoptosis (Figure 2).36 Pb also affects enzymatic reactions in which calcium plays a role, and the calcium-sensing receptor can also be activated by Pb, which suggests that there may be other mechanisms for lead nephrotoxicity.37,38
Pb induces activation of transcription nuclear factor kappa B, activation of the intrarenal renin-angiotensin system and attraction of macrophages, which generates an inflammatory process in the renal interstitium that may be involved in the development of tubulointerstitial damage and high blood pressure.39
In endothelial cells, it has been shown that increased formation of free radicals induced by Pb decreases nitric oxide production and the expression of the enzyme guanylate cyclase. These effects explain how high blood pressure can develop as a result of exposure to this metal.36-41 In addition, it stimulates the activity of NADP(H) oxidase by increasing production of hydrogen superoxide and hydrogen peroxide, thus affecting oxidative stress and the intracellular redox potential.42
Clinical manifestations
Acute exposure to high doses of Pb can cause PCT lesions, which manifest clinically as aminoaciduria, glycosuria or hyperphosphatemia. Other clinical manifestations include haemolytic anaemia, acute attacks of gout, intense abdominal pain (“painter’s colic”) and encephalopathy.43
Diagnosing chronic nephritis due to Pb is difficult, since urinary symptoms and findings are variable and lack specificity. Diagnosis is therefore based largely on a clinical history of exposure. Chronic exposure is associated with tubulointerstitial nephritis and progressive deterioration of renal function. Urinary excretion of urates decreases due to the effect of Pb on the PCT and renal blood flow decreases as well, resulting in increased urate levels in the bloodstream.44
In bone, chronic exposure is related to the pathogenesis and progression of osteoporosis, since Pb has adverse effects on osteoblasts and osteoclasts that affect bone formation and reabsorption.45
There is no adequate treatment for decreasing high Pb levels in the blood, but EDTA chelation therapy (1g in 200ml saline at 0.9%, administered weekly during 3 months) helps decrease Pb toxicity. Preventing exposure to this metal is the best means of reducing high levels in the bloodstream.35
ARSENIC NEPHROTOXICITY
Arsenic is one of the most widespread environmental pollutants and millions of people (mostly in Asia and Latin America) suffer from exposure to As, since it is a common pollutant in drinking water. Another less common form of exposure is through medications containing As, such as arsenic trioxide used in the treatment of acute promyelocytic leukaemia, and other drugs used to treat sleeping sickness and leishmaniasis.46
Epidemiology
The causal association between As and the formation of tumours in the skin, lungs, bladder, liver and kidneys has been exhaustively described. Some epidemiological studies have shown an association between exposure to high levels of As and increased risk of cardiovascular disease and diabetes mellitus. However, studies conducted in areas with low to moderate exposure have not yet demonstrated this association conclusively.
Until now, there have been few reports in the literature on the effects of As on the renal function of the general population. Hsueh et al47 studied 125 people with GFR<60ml/min and 229 people with normal renal function and found a weak association between urinary levels of As and decreased renal function (r2=0.04, P≤.001). Meliker et al48 showed that in patients with decreased renal function, higher As levels were associated with higher mortality rates (OR: 1.11, 95% CI: 1.09-1.13).
Physiopathology
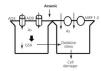
As is absorbed by the intestine, lungs (inhalation) and, to a lesser extent, through the skin. Once it has been absorbed, it is transported to all tissues in the body. The intake of selenium and vitamin B decrease intestinal absorption of As. Arsenic is methylated in the liver in a GSH-mediated process which decreases its toxicity and facilitates its biliary and urinary excretion.49 Arsenic enters the intracellular medium through the aquaglyceroporins AQ3 and AQ9 and studies in cell cultures have shown that the increase in AQ3 and AQ9 cellular expression increases intracellular accumulation of As. In the liver, AQ9 is important for biliary excretion of As.50
Another group of As transport proteins includes MRP-1 and 2 (ATP binding cassette-multidrug resistance protein) which were first described in the liver, where they transport As bound to GSH to the bile. The MRP-2 transporter is also located in proximal tubule cells, which favours entry of As into these cells.51 Arsenic toxicity in PCT cells is due to GSH depletion and an increase in oxidative activity by free radicals (Figure 3).
The literature does not currently offers sufficient information on clinical manifestations of As toxicity in the kidneys, but it is likely to manifest as data indicating tubular damage, such as low molecular weight proteinuria, aminoaciduria, glycosuria and phosphaturia, as well as progressive deterioration of renal function.52
CONCLUSION
In conclusion, there is ample evidence of the renal damage associated with these heavy metals. In addition, the combination of different metals has been shown to have a cumulative nephrotoxic effect. Since these metals are commonly found in the environment and there are no treatment options that decrease their systemic effects, increased vigilance is needed in order to decrease environmental levels of Pb, Cd and As.
Conflicts of interest
The authors declare potential conflicts of interest:
- Grants awarded: mixed funding from the State of Querétaro (Fondos Mixtos del Estado de Querétaro). Mexican National Council for Science and Technology.
KEY CONCEPTS

Figure 1. Physiopathological mechanisms of cadmium-induced kidney injury

Figure 2. Physiopathological mechanisms of lead-induced kidney injury

Figure 3. Physiopathological mechanisms of arsenic-induced kidney injury



