




To the Editor,
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterised by inflammation and fibrosis in the intrahepatic and extrahepatic bile ducts, which primarily affects middle-age males.1,2 It can occur as an isolated entity or in association with inflammatory bowel disease. The aetiopathogenesis of PSC has not been established, although growing evidence points towards genetic and immunological causes.1
Associated interstitial nephropathy in patients with chronic cholestatic liver disease was first described in the medical literature during the 1990s in paediatric patients, and it was suggested that this association might represent a new syndrome.3,4
Recently, the existence of a new disease, called “IgG4-related sclerosing disease” has been proposed.5,6
Here, we describe the case of a female patient diagnosed with PSC with severe interstitial nephropathy.
CASE REPORT
A 77 year-old female was referred to the nephrology department due to plasma creatinine values of 2.4mg/dl. The patient’s history included several surgical procedures: meniscus in 1985, thymoma with benign histology in 1999, nasal sinus polyps more than 20 years ago and again in 2008, varicose veins in 2006, cystocele in 2008, and hip fracture in July 2010. She was diagnosed with primary sclerosing cholangitis in 2003, and was under treatment with ursodeoxycholic acid. On several occasions, the patient had been placed retrograde biliary catheters and undergone sphincterotomy and dilation of the areas of stenosis. Inflammatory bowel disease had also been diagnosed around the same time. In later follow-up sessions, she underwent several colonoscopies, which occasionally produced normal results, and at other times revealed small ileocaecal and colonic ulcers. The patient also suffered an episode of pericarditis of unknown cause in January 2010, and had bilateral gonarthrosis. The patient was intolerant to oral iron supplements, and had no toxic habits.
At the first visit, the patient was taking zolpidem 10mg/day, ursodeoxycholic acid 1250mg/day, pantoprazole 40mg/day, mirtazapine 15mg/day, and occasional paracetamol and dextropropoxyphene.
The patient complained of intense fatigue, dyspnoea after moderate exercise, reduced appetite, occasional nausea associated with coughing, periodical constipation lasting 48 hours and alternating with diarrhoea that produced 3 or 4 evacuations per day, but with no pathological signs, nocturia twice per day for several months, diurnal urination every 3-4 hours, and no history of reno-ureteral lithiasis or haematuria.
As regards family background, the patient’s parents both died at an elderly age, and three brothers had died as a result of tumours.
The physical examination revealed the following values for standard parameters: height: 159cm; weight: 53kg; blood pressure: 137/72mm Hg; heart rate: 95bpm. We did not observe jugular vein engorgement, carotid pulses were rhythmic and symmetrical, cardiopulmonary auscultation normal, and we observed hepatomegaly of approximately 4 finger-widths in the right lobe, with no pain or abdominal murmurs. The limbs were without oedema, and distal pulses were present.
The patient’s plasma creatinine values were 2.4-2.5mg/dl in November 2010, 2.9mg/dl in December 2010, and 2.7mg/dl in January 2011.
Complementary tests in January and February 2011 revealed the following: cholesterol: 204mg/dl; glutamic-oxaloacetic-transaminase (GOT): 37U/l; glutamic-pyruvic-transaminase (GPT): 23U/l; gamma-glutamyl transferase (GGT): 431U/l; alkaline phosphatase: 483U/l; lactate dehydrogenase (LDH): 646U/l; ferritin: 399ng/ml; transferrin saturation index: 11%. All other biochemical parameters were normal. The results of a haemogram were: haematocrit: 35.1%; haemoglobin: 11.3g/dl, and all other values were normal. The results of a blood clotting test were: activated partial thromboplastin time: 45 seconds. The patient was anticardiolipin IgM positive: 21.80U MPL/ml (normal: 0.5-11).
The results of a 24-hour urine test were: proteinuria: 0.53g/24 hours; creatinine clearance: 17ml/min. The urine resulted negative.
An immunological analysis was positive for anti-neutrophil cytoplasmic antibodies (ANCA) (anti-MPO and anti-PR3 negative); the test was negative for anti-nuclear antibodies (ANA), anti-DNA antibodies, anti-smooth muscle antibodies, and anti-mitochondrial antibodies. We also detected a decrease in immunoglobulins: IgG: 253mg/dl (normal: 751-1560mg/dl); IgM: 10mg/dl (normal: 46-304mg/dl); IgA: 81mg/dl (normal: 82-453mg/dl). Complement, rheumatoid factor, C-reactive protein, and ceruloplasmin were all within normal ranges.
Serology for hepatitis B and C and HIV were all negative.
Thyroid hormones and anti-thyroid antibodies were within normal levels. Intact parathyroid hormone was 149pg/ml.
Electrophoresis revealed decreased immunoglobulins in a blood sample, and non-selective proteinuria in a urine sample. A Bence-Jones proteinuria test was negative.
In imaging tests (renal ultrasound), we observed kidneys with normal size, morphology, and echogenicity, with no lithiasis or dilation.
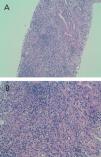
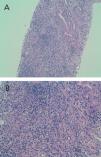
Given the deteriorated renal function of unknown cause (we had no previous reference values to compare with), we performed a percutaneous renal biopsy with the following findings: 4 glomeruli, 2 of which were completely sclerosed, and the other 2 progressing towards sclerosis. An immunofluorescence study was negative for IgG, IgA, IgM, and C3. We observed dense lymphocyte infiltration expanding throughout the interstitial tissue, destroying the normal architecture of the renal parenchyma (Figure 1A and Figure 1B). We could not properly assess fibrosis due to the high level of infiltration. Figure 2A displays the immunohistochemical results using CD3 (monoclonal antibody that marks all T-lymphocytes), demonstrating that the majority of the infiltration is due to T-lymphocytes, and Figure 2B shows the results of the immunohistochemical analysis using CD4. The final histological diagnosis was severe tubulointerstitial nephropathy (interstitial infiltration was predominantly by CD4-positive T-lymphocytes).
The biopsy findings indicated treatment with prednisone 50mg/day in a progressively decreasing prescription.
Eight months later, the patient has stable renal function (plasma creatinine: 2.2mg/dl; creatinine clearance: 20ml/min; proteinuria: 0.49g/24h). Current liver function values are: GOT: 24U/l; GPT: 18U/l; GGT:172U/l; alkaline phosphatase: 342U/l.
DISCUSSION
PSC is a disease of an unknown aetiology, with a progressive clinical presentation that affects the bile ducts and causes cirrhosis.1 Patients with PSC can have a number of serological findings, including hyper-gamma-globulinaemia and positive tests for atypical ANCA and other auto-antibodies such as anti-nuclear, anti-smooth muscle, anti-thyroid, anti-cardiolipin, and rheumatoid factor antibodies.7 Elevated IgG4 levels (a characteristic marker of autoimmune pancreatitis) have also been described in some patients with PSC.8 In fact, IgG4-related sclerosing cholangitis is also included in the group of idiopathic sclerosing cholangitis.5
Recently, “IgG4-related sclerosing disease” has been classified as a new pathological/clinical entity.5-7 It is a systemic disease characterised by infiltration of plasma IgG4-positive and T-lymphocyte cells into several different organs: pancreas, bile ducts, salivary glands, retroperitoneum, kidneys lungs, and prostate. The histological findings in affected organs include fibrosis, sclerosis, destroyed glandular architecture, and lymphoplasmacytic infiltration.
Tubulointerstitial nephropathy is also related to certain medications, infections, and autoimmune processes. Several cases of interstitial nephropathy have been associated with sclerosing cholangitis, leading to speculation about a new syndrome.
We have described a new case of interstitial nephritis in a female patient with PSC. In our case, we evaluated whether this could be in the context of IgG4-related sclerosing disease, but could not show such an association: firstly, because plasma IgG levels were 1mg/dl (although the patient did have an overall decrease in all immunoglobulins), and secondly, because the renal histological analysis did not report positivity for IgG4. However, the levels of IgG4 alone are insufficient to include or exclude the diagnosis of IgG4-related systemic disease.9 In fact, we observed dense interstitial infiltration of predominantly CD4-positive T-lymphocytes, which practically destroyed all renal structures.
There is no standardised treatment protocol for this new entity, although the majority of studies have prescribed 30-60mg/day of prednisolone in a progressively decreasing prescription, with good responses.5-7 In our case, the severe level of interstitial damage led us to indicate treatment with oral steroids: although we did not reach a complete recovery of renal function (possibly because the deterioration was already several months progressed before the biopsy, with severe histological damage), we did manage to stabilise renal function.
In conclusion, we have described the case of severe interstitial nephritis in a woman with primary sclerosing cholangitis: in these patients, we should examine whether this association could be referred to as an emerging pathology: “IgG4-related systemic disease.”
Conflicts of interest
The authors affirm that they have no conflicts of interest related to the content of this article.

Figure 1A. Haematoxylin eosin staining (x10): dense interstitial lymphocyte infiltration that destroyed most recognisable structures

Figure 1B. Haematoxylin eosin staining (x20): dense interstitial lymphocyte infiltration that destroyed most recognisable structures

Figure 2A. Immunohistochemical staining (x20) with CD3

Figure 2B. Immunohistochemical staining (x20) with CD4



