









Primary hyperoxaluria (PH) occurs due to an autosomal recessive hereditary disorder of the metabolism of glyoxylate, which causes excessive oxalate production. The most frequent and serious disorder is due to enzyme deficit of alanine-glyoxylate aminotransferase (PH type I) specific to hepatic peroxisome. As oxalate is not metabolised in humans and is excreted through the kidneys, the kidney is the first organ affected, causing recurrent lithiasis, nephrocalcinosis and early renal failure. With advance of renal failure, particularly in patients on haemodialysis (HD), calcium oxalate is massively deposited in tissues, which is known as oxalosis. Diagnosis is based on family history, the presence of urolithiasis and/or nephrocalcinosis, hyperoxaluria, oxalate deposits in tissue forming granulomas, molecular analysis of DNA and enzyme analysis if applicable. High diagnostic suspicion is required; therefore, unfortunately, in many cases it is diagnosed after its recurrence following kidney transplantation. Conservative management of this disease (high liquid intake, pyridoxine and crystallisation inhibitors) needs to be adopted early in order to delay kidney damage. Treatment by dialysis is ineffective in treating excess oxalate. After the kidney transplant, we normally observe a rapid appearance of oxalate deposits in the graft and the results of this technique are discouraging, with very few exceptions. Pre-emptive liver transplantation, or simultaneous liver and kidney transplants when there is already irreversible damage to the kidney, is the treatment of choice to treat the underlying disease and suppress oxalate overproduction. Given its condition as a rare disease and its genetic and clinical heterogeneity, it is not possible to gain evidence through randomised clinical trials. As a result, the recommendations are established by groups of experts based on publications of renowned scientific rigour. In this regard, a group of European experts (OxalEurope) has drawn up recommendations for diagnosis and treatment, which were published in 2012.
La hiperoxaluria primaria (HOP) se debe a un desorden metabólico hereditario autosómico recesivo, del metabolismo del glioxalato, que causa una producción excesiva de oxalato. El trastorno más frecuente y grave se debe al déficit enzimático de alanin:glioxalato aminotransferasa (HOP tipo I) específico en el peroxisoma hepático. Dado que el oxalato no se metaboliza en los humanos y se elimina por vía renal, el riñón es el primer órgano afectado, dando lugar a la aparición de litiasis de repetición, nefrocalcinosis e insuficiencia renal precoz. Con la progresión de la insuficiencia renal, especialmente en pacientes sometidos a hemodiálisis (HD), el oxalato cálcico se deposita masivamente en los tejidos, denominándose a esto último oxalosis. El diagnóstico se basa en los antecedentes familiares, la presencia de urolitiasis y/o nefrocalcinosis, hiperoxaluria, depósitos tisulares de oxalato formando granulomas en formas avanzadas, análisis molecular de ADN y análisis enzimático si procede. Se requiere una alta sospecha diagnóstica, por lo que, desafortunadamente, en muchos casos se diagnostica tras su recidiva en el trasplante renal. El manejo conservador de la enfermedad (alta ingesta líquida, piridoxina e inhibidores de la cristalización) debe ser precoz, para retrasar el daño renal. El tratamiento con diálisis es inefectivo para depurar el exceso de oxalatos. Tras el trasplante renal suele observarse una rápida aparición de los depósitos de oxalato en el injerto y los resultados de esta técnica, salvo excepciones, son desalentadores. El trasplante hepático anticipado, o simultáneo con el trasplante renal cuando ya existe daño irreversible de este órgano, es la opción terapéutica de elección para corregir la enfermedad de base y suprimir la sobreproducción de oxalatos. Dada la condición de enfermedad rara y su heterogeneidad genética y clínica, no es posible obtener evidencias a través de ensayos clínicos aleatorizados. Por lo tanto, las recomendaciones las establecen grupos de expertos apoyados en publicaciones de acreditado rigor científico. En este sentido, un grupo de expertos europeos (OxalEurope) ha elaborado unas recomendaciones diagnósticas y terapéuticas publicadas en 2012.
INTRODUCTION
Primary hyperoxaluria (PH) is an autosomal recessive hereditary glyoxylate metabolism disorder in which there is an excessive production of oxalate. The most common disorder is due to a deficiency of the enzyme alanine: glyoxylate aminotransferase (PH type I), which is specific to hepatic peroxisome1-4. The incidence of PH is difficult to estimate, given that many cases are identified late or are never identified. It has an estimated prevalence of 1-3 per million population and an incidence rate of approximately 1:100,000 births5,6. Higher rates have been reported in historically isolated populations, such as in the Canary Islands, due to a founder effect7. It affects at least 1% of the paediatric population with end-stage renal disease and it is more common in populations where consanguinity is higher8.
OXALIC ACID METABOLISM
Oxalate is a dicarboxylic acid (C2O4H2, molecular weight 90Da) that mainly comes from endogenous metabolism and only a small part comes from the diet. It is produced in the liver from glyoxylate. It is a molecule generated in the intermediate metabolism of glycine, hydroxyproline and glycolate. The detoxification of glyoxylate is mostly carried out by alanine-glyoxylate aminotransferase (AGT) in the human hepatocyte peroxisome, which converts glyoxylate into glycine. Vitamin B6 acts as a cofactor. In normal conditions, only part of the glyoxylate is transformed into oxalate by lactate dehydrogenase (LDH)9 (Figure 1). Oxalate cannot be metabolised by mammals, it does not bind to proteins and it is not metabolised. It is filtered by the glomerulus and is also secreted by the tubule, being eliminated unchanged via the kidneys. Urinary elimination is normally less than 45mg/day or 0.45mmoles/l/1.73m2 per day (mg are converted to mmoles by multiplying by 0.01136)4,10,11.
The genetic defects of enzymes that metabolise glyoxylate lead to overproduction of oxalate in the liver; that is, this disease is due to excess production. This is called PH and the term oxalosis applies to tissue deposits.
CLASSIFICATION OF HYPEROXALURIC STATES
The main and most serious cause of hyperoxaluria are hereditary enzyme defects known as PH. In these cases, urinary excretion of oxalate is >45mg/day/1.73m2 with a frequency >801,12. Furthermore, there are other situations called secondary hyperoxalurias (SHO)13. The causes of SHO are the abusive intake of oxalate precursors and an increase in intestinal absorption (enteric hyperoxaluria). In general, these are less severe forms (<70mg/dl) and do not usually result in systemic oxalosis. Table 1 displays the classification of hyperoxaluric states.
Primary hyperoxaluria
This is an autosomal recessive hereditary glyoxylate metabolism disorder, which is the result of a high production of oxalate in the liver2,14,15. Three types of molecular disorders have been reported. The genes involved are alanine-glyoxylate aminotransferase (AGXT) for type 1 PH, (PH-I), representing 80% of patients who have PH, glyoxylate reductase/hydroxypyruvate reductase (GRHPR), located on chromosome 10, for PH-II, and 4-OH-2-oxoglutarate aldolase (HOGA1, also known as DHDPSL), located on chromosome 9, for PH-III16.
Primary hyperoxaluria type I
PH-I is caused by a deficiency of AGT, an enzyme that catalyses the transamination of L-alanine and glyoxylate to pyruvate and glycine. It is specifically expressed in the liver of humans and is located in the peroxisome4,17-20. Its deficiency causes an increase in glycolate and oxalate21. Cloning of AGXT cDNA and knowledge of its three-dimensional structure have provided important information about the protein’s functions and the effect of changes in the amino acid sequence in most of the more than 150 mutations reported in PHO22-24.
These mutations result in different phenotypic expressions of the disease due to various molecular mechanisms4. Four basic mechanisms have been reported:
Molecular mechanism research will probably lead to a better knowledge of genotype-phenotypic correlations and to research of new therapeutic options.
Primary hyperoxaluria type II
PH-II or L-glyceric aciduria represents around 10% of PH. Deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR) induces increased urinary excretion of oxalate and L-glycerate. These patients have a less severe urinary oxalate increase than in PH-I and few develop end-stage renal disease30.
Primary hyperoxaluria type III
It occurs in 10% of PH cases; mutation of the HOGA1 gene causes deficiency of the mitochondrial enzyme 4-OH-2-oxoglutarate aldolase, which divides 4-OH-2-oxoglutarate into pyruvate and glyoxylate, which in turn is transformed into oxalate by LDH. It occurs with a wide range of urinary oxalate elimination values, but progression to end-stage renal disease has not been reported16.
Secondary hyperoxaluria
The causes of SHO are summarised in Table 1.
Enteric hyperoxaluria
Oxalate from the diet may be absorbed along the gastrointestinal tract, both by passive diffusion and by active transport. All situations that cause malabsorption of bile salts and fatty acids in the ileum may increase oxalate absorption31. This is because upon reaching the colon, they increase the permeability of the colonic mucosa to oxalate, and furthermore calcium binds to fatty acids, leaving oxalate free and soluble and therefore, easily absorbable32-36. Experimental studies have also demonstrated that the acidification of the colon with lactulose significantly increases urinary oxalate excretion37.
As such, inflammatory bowel diseases that occur with steatorrhea31,35 and extensive ileal resections38-41 may develop hyperoxaluria and lithiasis whenever the colon is intact35. The increased prevalence of renal lithiasis in cases of pancreatic cystic fibrosis, as well as hypocitraturia, has been related to an increased excretion of renal oxalate. Anecdotally, cases have been reported of renal graft loss due to oxalosis in transplant patients who developed steatorrhea secondary to biliopancreatic disorders43.
Hyperoxaluria and nephrolithiasis have more recently been reported as complications in patients who have undergone bariatric surgery to treat morbid obesity44-46, and even required chronic dialysis in two cases46. Therefore, regular monitoring of oxaluria is recommended in the early postoperative stages and in the long term in these patients.
Another important factor that may influence oxalate absorption is the decolonisation of oxalate metabolising bacteria in the colon, with Oxalobacter formigenes being the best studied (see below)47. The absence of these bacteria causes hyperoxaluria due to an increase in oxalate absorption. Antibiotic use has been suggested as a reason for its eradication.
Abusive intake of precursors
The intentional or accidental intake of ethylene glycol, which is most commonly used as an engine coolant, may cause a severe hyperoxaluric crisis48. The toxicity of ethylene glycol is related to its biotransformation to glycolic acid, causing severe normochloraemic acidosis, hyperoxaluria and oxalosis49,50.
Ascorbic acid (vitamin C) is a metabolic precursor of oxalate51, and it has been demonstrated that its abuse may induce calcium oxalate oversaturation and the complications resulting from its precipitation52-54.
CLINICAL PRESENTATION
There is considerable heterogeneity in the presentation pattern of the disease and its progression to renal failure55. This great clinical variability is not related to gene mutations or to the degree of residual enzyme activity. g Furthermore, there are factors that inhibit and promote oxalate crystallisation and modulate the genetic pattern, which favours wide clinical heterogeneity.
The most severe forms occur in HPO-I, with childhood forms being reported that appear in the first months of life, for which there is a high early mortality rate. The most common forms appear around the second decade of life and many patients are treated as having recurrent lithiasis, with the condition going years undiagnosed. In fact, HPO is frequently (30%-60%, according to the series) diagnosed after the recurrence of oxalate deposits in the kidney transplant3,7,56,57. This is even more unfortunate whenever the donor is a living donor, as occurred in one case in our patient series7. There are less aggressive variants that are diagnosed in adulthood due to the presence of lithiasis and/or nephrocalcinosis, which have long-term survival, even when the patient is on dialysis. There are even elderly patients who do not develop end-stage renal disease2,3.
In cases of HPO-II, the disease is usually less aggressive58 and only a small proportion of patients (approximately 20%) develop end-stage renal disease. Cases of HPO-III are even less common (10%) and more benign, although the experience is more limited in these patients16,59.
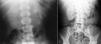
Recurrent nephrolithiasis, nephrocalcinosis, haematuria, urinary infections and rapid development of renal failure are prominent clinical manifestations. It should be highlighted that nephrocalcinosis is typical and is accompanied by nephrolithiasis or appears on its own (Figure 2).
Although this is a disease with a well-defined identity, given that it is uncommon, in patients with recurrent nephrolithiasis the doctor responsible usually studies the causes of SHO and the most common causes of nephrolithiasis. The differential diagnosis must be based on the following premises:
- Questions about and analysis of potential causes of SHO: intake of glyoxalate precursors and inflammatory bowel diseases or extensive intestinal resections, including bariatric surgery.
- Metabolic lithiases: hypercalciuria (>4mg/kg/day without hypercalcaemia or a glomerular filtration rate [GFR] >0.15mg/dl), hypocitraturia (<300mg/24 hours, or a GFR <0.17mg/dl) and hyperuricosuria (>750-800mg/24 hours or a GFR >0.45mg/dl). They do not occur with nephrocalcinosis or develop early renal failure.
- Hypercalcaemic states: sarcoidosis, primary hyperparathyroidism, hypervitaminosis D and milk-alkali syndrome. They may occur with lithiasis and nephrocalcinosis, but their differentiating feature is obviously hypercalcaemia, which does not appear in PH.
- Type 1 renal tubular acidosis: it may develop lithiasis and nephrocalcinosis if it occurs with hypercalciuria. However, its hallmark is hyperchloraemic metabolic acidosis and hypokalaemia and the development of renal failure is uncommon.
- Cystinuria: less radiopaque characteristic hexagonal crystals in the sediment do not develop nephrocalcinosis and the occurrence of advanced or end-stage renal disease is uncommon.
Lastly, on the basis of clinical data, Table 2 displays the clinical situations in which PH diagnosis must be studied, always in the absence of data that may suggest enteric hyperoxaluria or abnormal precursor intake.
Tissue damage in primary hyperoxaluria: oxalosis
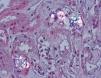
The kidney is the first organ affected, with calcium oxalate aggregates in the urinary space (urolithiasis) and in the renal tissue (nephrocalcinosis) (Figure 3), where there is significant development of interstitial fibrosis and renal failure1,60,61. Once the GFR falls below 30-40ml/min/1.73m2, urinary elimination is not capable of maintaining oxalaemia within normal levels (<6μmol/l) and the calcium oxalate saturation threshold may be exceeded whenever levels are greater than 30μmol/l, causing tissue deposits in the form of monohydrates and dihydrates62-64. This phenomenon is called oxalosis and produces a significant inflammatory response in the form of granulomas around the crystals.
Extrarenal calcium oxalate deposits have been identified in most tissues and organs, such as the retina, the cardiac muscle, blood vessels, skin, bone and the nervous system3,61,62,65, although they will preferentially occur where the calcium concentration is greatest. The development of cardiomyopathy and conduction disorders, vascular disease with frequent distal necroses66, retinopathy, synovitis or high remodelling bone disease65 are severe late complications that lead to early mortality.
Once on haemodialysis, tissue oxalate deposits progress spectacularly, and bone tissue is the most affected organ65,67. Patients develop progressive bone pain, disability and skeletal deformities. X-rays show a marked increase in bone density, bones lose their normal structure and osteitis fibrosa cystica type high remodelling lesions are observed, but of an unusual severity65,68.
The study and follow-up with a bone biopsy allowed us to find the starting point of the bone disease and follow its progression in haemodialysis65. In the pre-dialysis phase, clinical or radiological manifestations of the bone disease are absent, although emerging peritrabecular oxalosis foci may appear in the histological study (Figure 4A). This appears to be the initial deposit site, probably because it is the area with the highest calcium concentration. After one or two years, the lesions develop with unusual severity. There is evidence that the reaction of cells to crystal deposits is one that activates and accelerates bone remodelling, simulating hyperparathyroidism. Giant multinucleated cells that engulf the crystals belong to the macrophage-monocyte series, as well as osteoclasts. The action of this cell group on bone trabeculae adjacent to granulomas would trigger bone resorption. Due to the normal osteoclast-osteoblast coupling, the latter are activated generating a predominantly non-laminar osteoid. The dynamic studies reveal diffuse uptake of tetracyclines in non-laminar osteoid areas. The crystals mainly appear in the medullary space and invade the trabecular area. They are grouped together in the shape of a star or rosette and around them there is a granulomatous reaction to foreign bodies with giant multinucleated cells, macrophages, fibroblasts and medullar fibrosis (Figure 4B). On examination with polarised light, these crystals are highly refractive, appearing fragmented in subunits (Figure 4C)65. Serum levels of alkaline phosphatase and parathyroid hormone usually appear to be only slightly increased, in contrast with cases of severe hyperparathyroidism.
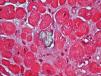
We also documented oxalate deposits in other tissues. In one patient with PH and severe polyneuropathy, by electron microscopy, we observed oxalate deposits in Schwann cells and in the myelin sheaths of the peripheral nerve a few months after starting dialysis (Figure 5). In another patient with heart disease, the diagnosis was made due to the presence of oxalate crystals in the subendocardial biopsy (Figure 6), which was later confirmed with DNA analysis. In two other PH patients on haemodialysis, we observed a primary and absolute resistance to treatment with erythropoietin, despite the use of high doses of this hormone. Severe bone oxalosis causes extensive bone marrow occupation by granulomatous tissue and fibrosis that affects erythropoiesis by replacing haematopoietic tissue.69,70 However, organs such as the liver were not affected, probably because their intracellular calcium concentration was lower.
DIAGNOSTIC METHODS
Early diagnosis is crucial for preventing end-stage renal disease. As we mentioned previously, a thorough family history, along with oxaluria tests, is essential in suspected cases of PH. Sediment tests may provide additional information with the presence of monohydrate calcium oxalate crystals (whewellite, CaCO4.H2O) in the shape of “dumbbells”, which must be distinguished from the typical dihydrate calcium oxalate diamond crystals71. If the diagnostic suspicion persists, testing glycolate in 24-hour urine and oxalaemia may provide additional information.
Although oxalate deposits in tissue usually occur later, the effects on the heart (electrocardiogram and echocardiogram), the fundus of the eye and the skeleton should be studied55. Computerised tomography may be very useful for assessing the extent of calcifications and tissue oxalate deposits.
Histological studies that demonstrate the presence of oxalate crystals, especially renal biopsies, may be very useful in uncertain cases.
Genetic analysis is necessary to confirm the diagnosis and categorise the mutational variant. Given that only DNA of peripheral blood is required, the study of common mutations has become the next diagnostic step, once hyperoxaluria has been observed. The enzyme activity of AGT in liver tissue may be useful if the genetic study is inconclusive72. Genetic advice and prenatal diagnosis by mutational analysis of the parents are appropriate recommendations.
A PH diagnosis algorithm was recently published by the European guidelines73. Figure 7 displays a simplified diagnostic algorithm guide, which should be adjusted according to individual clinical and demographic circumstances and the tools available in the setting.
Biochemical methods
Testing oxalate in urine is the first step in diagnosing PH. Oxaluria higher than 45mg/day (>0.5mmol/1.73m2/day) in at least two 24-hour urine samples is typical of PH, once causes of SHO have been ruled out1,12. Samples greater than 80-90mg/day are very suggestive of PH-I55.
Increased levels of glycolate is typical of PH-I and, although it has a low diagnostic specificity and sensitivity, urinary glycolate values greater than 45mg/day (>0.5mmol/1.73m2/day) are suggestive of PH-I12,55,74.
Increased levels of L-glycerate is indicative of PH-II. The extreme rarity of PH-II forms means that glycerate in urine can only be tested in hospitals that are highly specialised in the analysis of organic acids75.
In infants and young children, in whom urine collection is difficult, an isolated sample in the morning may be a good guide. These results must be interpreted in accordance with reference values for the patient’s age1,75,76.
Serum oxalate values remain within the normal range until renal filtration falls below approximately 45ml/min. Given the wide range of values, there is no reliable threshold for establishing the diagnosis. For values higher than 50µMol/l, the disease must be studied and values greater than 100µMol/l are very suggestive of PHO55,62,76,77.
Histological diagnosis
An abundance of oxalate crystals in the renal biopsy is indicative of PH, particularly if they appear in the interstitium, surrounded by a strong inflammatory reaction in the form of foreign body granulomas. It is not uncommon for these deposits to appear in a transplanted kidney and for them to provide a late diagnosis of the underlying disease7. In advanced cases of dialysis patients with high-remodelling bone disease images, the bone biopsy has provided the definitive diagnosis, showing the typical granulomatous reactions surrounding oxalate deposits7,65.
DNA analysis
The diagnosis of PH must be confirmed with a DNA analysis of the AGT gene (AGXT), which furthermore provides information about the type of mutation and polymorphisms affecting patients18,20,23,78-80. When there are potential non-common mutations, genetic analysis of immediate family members is necessary. Given that DNA analysis is a non-invasive method, it has become a preferred diagnostic test when there is a clinical and biochemical suspicion of PH, and is also used for the prenatal test and diagnosis of other family members once the mutation is known.
Enzyme diagnosis
A liver biopsy is indicated when no mutation is found in the AGXT, GRHPR or HOGA1 genes, in order to completely rule out the known variants of PH. The levels of AGT enzyme activity in PH-I patients is quite variable, particularly in those affected by the mistargeting mutation, where AGT is present in the mitochondria. In this case, immunohistochemical analysis may provide the enzyme’s subcellular location81,82.
Prenatal diagnosis
Given that AGT is only expressed in the liver, enzyme tests carried out in the amniotic fluid are not useful. However, through DNA analysis in pregnant women and relatives, we can know the prenatal diagnosis of the most common mutations83. This is the most useful diagnostic procedure, since it makes analysis of the AGXT gene possible from amniotic fluid cells or by biopsy of chorionic villi. It is likely that these molecular techniques will become the main PH diagnostic method, since it can be carried out with DNA from any body cell and provides less ambiguous information than determining enzyme activity in a liver biopsy.
TREATMENT
General measurements
Treatment must be introduced at an early stage and it is aimed at decreasing urinary saturation of calcium oxalate by increasing fluid intake and using urinary crystallisation inhibitors84. Fluid intake must be greater than 3l/m2/day85 and urinary pH must be maintained between 6.2 and 6.8. These general measurements are applicable to all hyperoxaluric states and their effectiveness will depend on the extent of the problem. As such, in severe cases of PH, it is usually very limited55.
The recommended crystallisation inhibitor regimens are as follows:
- Potassium citrate: it forms complexes with calcium, decreasing calcium oxalate precipitation and it increases urinary pH86. A daily potassium citrate dose of 0.1-0.15g/kg is recommended12,55,86,87. In cases of renal failure, potassium citrate must be replaced by sodium citrate88. Thiazides (in combination with potassium citrate) may be a useful supplement for reducing calciuria and increasing urinary volume89.
- Orthophosphate: it can be administered in doses of 30-40mg/kg/day90.
- Magnesium: it is a known inhibitor of mineralisation and furthermore, it reduces oxalate absorption when it is administered along with food. The recommended dose91 is 500mg/day/m2.
The use of vitamin D analogues may have an adverse effect on these patients, due to an increase in calcium absorption and, as a result, they may cause an oversaturation of calcium oxalate92.
A gastrostomy tube may be required in infants and small children to achieve these objectives55.
Diet
The reduction of oxalate intake is not very useful in PH, given that the endogenous oxalate source remains; however, it may be more useful in cases of enteric hyperoxaluria. The foods richest in oxalate are: nuts, plums, chocolate, tea, Coca-Cola, beetroot, strawberries, etc.
Calcium intake must not be restricted, since intestinal oxalate absorption increases as a result of it93.
The excessive intake of vitamin C must be avoided, particularly in dialysis patients, since ascorbic acid may be metabolised directly to calcium oxalate52.
Oxalate-degrading enzymes
Although mammals cannot metabolise oxalate, other living beings have enzymes such as oxalate oxidase and oxalate decarboxylase, which are capable of degrading it and they may provide a new solution for preventing the accumulation of oxalate in PHO94,95. In this regard, intestinal colonisation with Oxalobacter formigenes, a bacteria that metabolises oxalate, has shown promising results in rat models with PH and in some pilot studies on patients47,96.
For patients with intestinal flora disorders, the use of probiotics (food with living microorganisms added that remain active in the intestine) may be beneficial. These bacteria use intestinal oxalate as a source of energy and prevent their absorption. A combination of lactic acid-producing lactobacilli has been used in patients with enteric hyperoxaluria, both inflammatory and secondary to intestinal resections, with a reduction in oxaluria and oxalocalcic saturation being demonstrated97,98 . In rat models with high oxalate diets, Oxalobacter formigenes supplements significantly reduced oxaluria. It has also been observed that Oxalobacter formigenes interacts with the colonic mucosa, favouring excretion/secretion of oxalate from the blood to the intestine.99
Pyridoxine
Pyridoxine (vitamin B6) is perhaps the only method capable of effectively reducing oxalate production, but it is only applicable to cases of PH-I. Pyridoxal-5-phosphate is a form of vitamin B6 that acts as an AGT cofactor, increasing transamination of glyoxylate (oxalate precursor) to glycine. The recommended dose is 5mg/kg/day up to a maximum of 20mg/kg/day100. However, the safety of these doses is not well-known and cases of sensory neuropathy have been reported, and as such, it is recommended not to exceed 1g/day in adults and correct this dose for children and infants. Response is considered to be a >30% decrease in oxaluria after three months of treatment at the maximum dose2,3 . Milliner et al.90 reported the efficacy of long-term treatment with orthophosphate and pyridoxine, reducing urinary crystallisation of calcium oxalate in 25 patients with PH and maintained renal function. 75% of patients remained without dialysis 20 years after treatment, with renal function having decreased by an average of 1.4ml/min/1.73m2 per year. From the molecular perspective, there is a patient subgroup who carry one or two copies of G170Arg and Phe152Ile, whose mutation leads to the abnormal mistargeting of AGT to the mitochondria and which has demonstrated a better response to pyridoxine101-104.
Surgical management of urolithiasis
Management of lithiasis in PH involves the normal concomitant presence of nephrocalcinosis. Treatment with lithotripsy involves the risk of applying shock waves to areas of nephrocalcinosis. As such, endoluminal endoscopic management is usually the treatment of choice, while open surgery is exceptional in this field105,106.
Dialysis
Once renal failure has been established, all the general measures mentioned are usually ineffective and renal replacement therapy must be planned. Haemodialysis and, even more so, peritoneal dialysis have demonstrated that they are ineffective at clearing oxalate generated in PHO107,108 . Oxalate is a small molecule, easily filtered, but the quantity of oxalate produced by the liver in PH may be significantly higher (350-600 mg/dl per day) than the clearance capacity of conventional dialysis (80-180 mg/dl in adults), resulting in a daily calcium oxalate deposit of 180-360mg/day63,77,107-110. Ideally, serum levels should be maintained below 30µMol/l62. From these studies, it appears that in order to achieve oxalate balance in haemodialysis, the sessions should be extended from 13 to 15 hours, which is impractical. Therefore, dialysis must be used while the patient is waiting for a transplant, with high-efficacy and intensive dialysis protocols being applied7,111.
Renal transplantation
The isolated use of renal transplantation has yielded unfortunate results. After implantation, the recurrence of nephrocalcinosis is normal, especially in cases of delayed renal function or episodes of rejection, and as such, we should consider it as a temporary or maintenance solution while liver transplantation is being planned1,7,8,84,112,113. PH has frequently been diagnosed after renal transplantation, with the appearance of oxalate deposits and an early loss of graft for this reason. These situations have even been documented in unfortunate cases of living donors, where PH had not been diagnosed7.
Therefore, given the high risk of rapid development of nephrocalcinosis, isolated renal transplantation should be reserved for less severe forms of PH, with a reasonable good response to conservative measures and vitamin B6. In these cases, renal failure appears in older age and progresses very slowly. Early transplantation (in the “pre-dialysis” phase) is a difficult decision, given the uncertainty of renal damage progression, with a better option being to schedule it as soon as possible after dialysis has begun. The living donor option is clearly advised against given the poor prognosis of isolated renal transplantation, although it may be considered in very specific exceptional circumstances.
The occurrence of nephrocalcinosis in a functioning graft is due to the rapid liberation of systemic calcium oxalate deposits. As such, it is key to apply an aggressive pre- and immediate post-transplant protocol, maximising the immunosuppressant protocol, minimising cold ischaemia time and maintaining fluid diuresis. Likewise, early and intensive (daily) dialysis support should be provided and all the anti-lithogenic measures described should be introduced (pyridoxine, crystallisation inhibitors and thiazide diuretics)7,114. During the subsequent follow-up, regular monitoring of oxaluria and potential tissue oxalate overload with diagnostic imaging techniques is essential.
Liver and renal transplantation
Once there is a firm diagnosis of PH, the priority and potentially curative treatment is liver transplantation7,115. Given that the normal situation is advanced and irreversible renal deterioration, the indication is dual liver and renal transplantation.1 The first combined liver and renal transplantation successfully carried out for treating PH was published by Watts et al. in 1987116. Since then, a considerable number of publications have demonstrated that simultaneous liver and renal transplantation is the treatment of choice in patients with advanced renal damage, even in infants7,84,117-121. Data obtained from the European Transplant Registry on PH demonstrated a good patient survival rate: 80% after 5 years and 68% after 10 years119. It should be highlighted that transplantation should be scheduled early, with a GFR between 15 and 30ml/min/1.73m2, in order to avoid oxalate tissue deposits.
Other transplantation alternatives have also been reported for special situations. Split living donor liver donation involves risks for the donor, and the amount of liver transplanted may be insufficient for preventing renal oxalate overload from the remaining liver. This option is currently advised against, being reserved only for exceptional circumstances.
The sequential transplantation alternative requires two donors, but it is less aggressive from the surgical perspective. If the GFR is higher than 15-20ml/min, the option is liver transplantation first and subsequent renal assessment. If the GFR is <15-20ml/min, the option is renal transplantation first. This may be considered in cases of advanced renal damage, but when progression has been very slow, normally in patients older than 50-60 years of age.
Early liver transplantation
The rapid progression of oxalosis, once renal failure has been established, indicates that liver transplantation should be performed prior to the development of the latter, thus avoiding the need for dual transplantation. Early liver transplantation, that is, before there is irreversible kidney damage, should be the option of choice in severe cases if we have an accurate and early diagnosis7,122-126. This alternative would have to be applied with renal function between 20 and 50ml/min115. Likewise, based on previous considerations about serum oxalate levels and their saturation threshold, patients with oxalaemias >50µmol/l if the GFR is <20ml/min, or >20µmol/l if the GFR is <30ml/min, should be considered potential candidates for liver transplantation62,64.
Table 3 summarises the PH transplantation methods and potential indications are suggested.
Molecular therapy prospects
Recent advances in the molecular mechanisms of the disease have made the development of animal genetic models and in vivo experimentation of effective and non-invasive therapeutic approaches possible.
Genetic therapy
Hereditary diseases such as PH may be resolved by providing the normal gene sequence to defective cells. The problem would be that hepatocytes not reached would continue generating oxalate, thus overloading the kidneys. The development of viral vectors with high hepatic tropism and a low immune response may contribute to genetic therapy being a realistic option for PH in the future128.
Chemical “chaperones” and therapy for regulating “proteostasis”
The term “proteostasis” is used to define homeostasis of intracellular proteins. Therefore, it combines concepts of folding and protein stability, degradation, intracellular traffic and expression of protein regulation systems. This complex and highly regulated network determines the intracellular destination of proteins129. Recent studies have demonstrated that small organic components called “chaperones”, may be involved in protein folding abnormalities, mitochondrial mistargeting and AGT aggregation and degradation processes23,78,130, resulting in PH presentation variants.
Cell transplantation
Partial liver transplantation failed to provide a sufficient reduction in oxalate production to prevent PH-I119. However, liver cell transplantation is a promising alternative, which is being studied in liver metabolism genetic defects including PH-I131,132. Transplantation of hepatocytes is a minimally invasive procedure, although the number of healthy hepatocytes injected in a single session is not sufficient for correcting a high oxalate production by native hepatocytes. In our experiments with mice models with PH-I, we calculated that approximately 40% of hepatocytes need to be transferred in order to reverse the hyperoxaluric phenotype128.
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.
Acknowledgements
This study was financed by: PROYECTO FIS PI070963 (Instituto de Salud Carlos III, and Fondos FEDER), RedInRen RD12/0021/0008, and SAF2011-23933 (MINECO project). One author (A. Torres) is a member of RedInRen (Red de Investigación Renal). One author (E Salido) is a member of CIBERER (Centro de Investigación Biomedica en Red de Enfermedades Raras).

Table 1. Classification of hyperoxaluric states.

Table 3. Transplantation methods in primary hyperoxaluria and possible indications.

Figure 1. Oxalic acid metabolism.

Figure 2. Multiple renal lithiases and nephrocalcinosis in patients with primary hyperoxaluria. Simple abdominal x-ray.

Figure 3. Calcium oxalate deposits in the urinary space and in the renal tissue (HE, polarisation microscope, 200X).

Figure 4. Bone oxalosis.

Figure 5. Oxalate deposits in Schwann cells.

Figure 6. Oxalate deposits in subendocardial tissue (HE, polarisation microscope, 20X).

Figure 7. Primary hyperoxaluria diagnostic algorithm.

Table 2. Clinical parameters in which the diagnosis of primary hyperoxaluria must be studied.



