





Mitogen-activated protein kinases (MAP kinases) are functionally connected kinases that regulate key cellular process involved in kidney disease such as all survival, death, differentiation and proliferation. The typical MAP kinase module is composed by a cascade of three kinases: a MAP kinase kinase kinase (MAP3K) that phosphorylates and activates a MAP kinase kinase (MAP2K) which phosphorylates a MAP kinase (MAPK). While the role of MAPKs such as ERK, p38 and JNK has been well characterized in experimental kidney injury, much less is known about the apical kinases in the cascade, the MAP3Ks. There are 24 characterized MAP3K (MAP3K1 to MAP3K21 plus RAF1, BRAF and ARAF). We now review current knowledge on the involvement of MAP3K in non-malignant kidney disease and the therapeutic tools available. There is in vivo interventional evidence clearly supporting a role for MAP3K5 (ASK1) and MAP3K14 (NIK) in the pathogenesis of experimental kidney disease. Indeed, the ASK1 inhibitor Selonsertib has undergone clinical trials for diabetic kidney disease. Additionally, although MAP3K7 (MEKK7, TAK1) is required for kidney development, acutely targeting MAP3K7 protected from acute and chronic kidney injury; and targeting MAP3K8 (TPL2/Cot) protected from acute kidney injury. By contrast MAP3K15 (ASK3) may protect from hypertension and BRAF inhibitors in clinical use may induced acute kidney injury and nephrotic syndrome. Given their role as upstream regulators of intracellular signaling, MAP3K are potential therapeutic targets in kidney injury, as demonstrated for some of them. However, the role of most MAP3K in kidney disease remains unexplored.
Las proteínas quinasas activadas por mitógenos (MAP quinasas) son quinasas conectadas funcionalmente que regulan procesos celulares clave involucrados en la enfermedad renal como la supervivencia, la muerte, la diferenciación y la proliferación. El típico módulo MAP quinasa está compuesto por una cascada de 3 quinasas: una MAP quinasa quinasa quinasa (MAP3K) que fosforila y activa una MAP quinasa quinasa (MAP2K) que, a su vez, fosforila una MAP quinasa (MAPK). Si bien el papel de las MAPK como ERK, p38 y JNK se ha caracterizado bien en las lesiones renales experimentales, se sabe mucho menos acerca de las quinasas apicales en la cascada, las MAP3K. Hay 24 MAP3K (MAP3K1 a MAP3K21, más RAF1, BRAF y ARAF). En este trabajo revisamos el conocimiento actual sobre la participación de MAP3K en la enfermedad renal no maligna y las herramientas terapéuticas disponibles. Existe evidencia intervencionista experimental in vivo que respalda claramente el papel de MAP3K5 (ASK1) y MAP3K14 (NIK) en la patogenia de la enfermedad renal experimental. De hecho, el inhibidor de ASK1, selonsertib, ha sido estudiado en ensayos clínicos en la enfermedad renal diabética. Además, aunque la MAP3K7 (MEKK7, TAK1) es necesaria para el desarrollo renal, la inhibición de MAP3K7 en el adulto protegió de la lesión renal aguda y crónica experimental; e inhibir MAP3K8 (TPL2/Cot) protegió de la lesión renal aguda. Por el contrario, MAP3K15 (ASK3) puede proteger de la hipertensión y los inhibidores de BRAF, en uso clínico, pueden inducir lesión renal aguda y síndrome nefrótico. Dado su papel como reguladores de los primeros pasos de la señalización intracelular, las MAP3K son posibles dianas terapéuticas en la lesión renal, como se ha demostrado para algunas de ellos. Sin embargo, el papel de la mayoría de las MAP3K en la enfermedad renal no ha sido explorado.
Chronic kidney disease (CKD) and acute kidney injury (AKI) are major causes of death and disability.1,2 It is estimated that 10% of the adult population worldwide has CKD while the incidence of AKI is growing.3,4 Both are associated with an increased risk of premature death and, in fact, CKD was among the fastest growing causes of death worldwide, according to the Global Burden of Disease (GBD) study.5 As an example, data from the GBD 2016 Spain analysis suggest that at the current rate of growth, CKD will become the second most common cause of death, after Alzheimer, by the year 2100.6,7 This growing burden of kidney disease reflects the need for earlier biomarkers of kidney injury that allow preventive rather than therapeutic intervention as well as the need for more effective therapeutic interventions.2,8 Kinases are a family of intracellular signaling regulators that have become therapeutic targets for diverse diseases and kinase inhibitors are in clinical use mainly in the oncology and immune regulation fields.9 Among kinases, mitogen-activated protein kinase kinase kinases (MAP3K) are of special interest given their localization at the apex of diverse intracellular signaling pathways in response to environmental stimuli and recent preclinical evidence supporting a key role of some of them, such as MAP3K14, in the pathogenesis of kidney injury.10 We now review MAP3Ks and the preclinical and clinical evidence suggesting a contribution to kidney injury and that they may become therapeutic targets in both AKI and CKD.
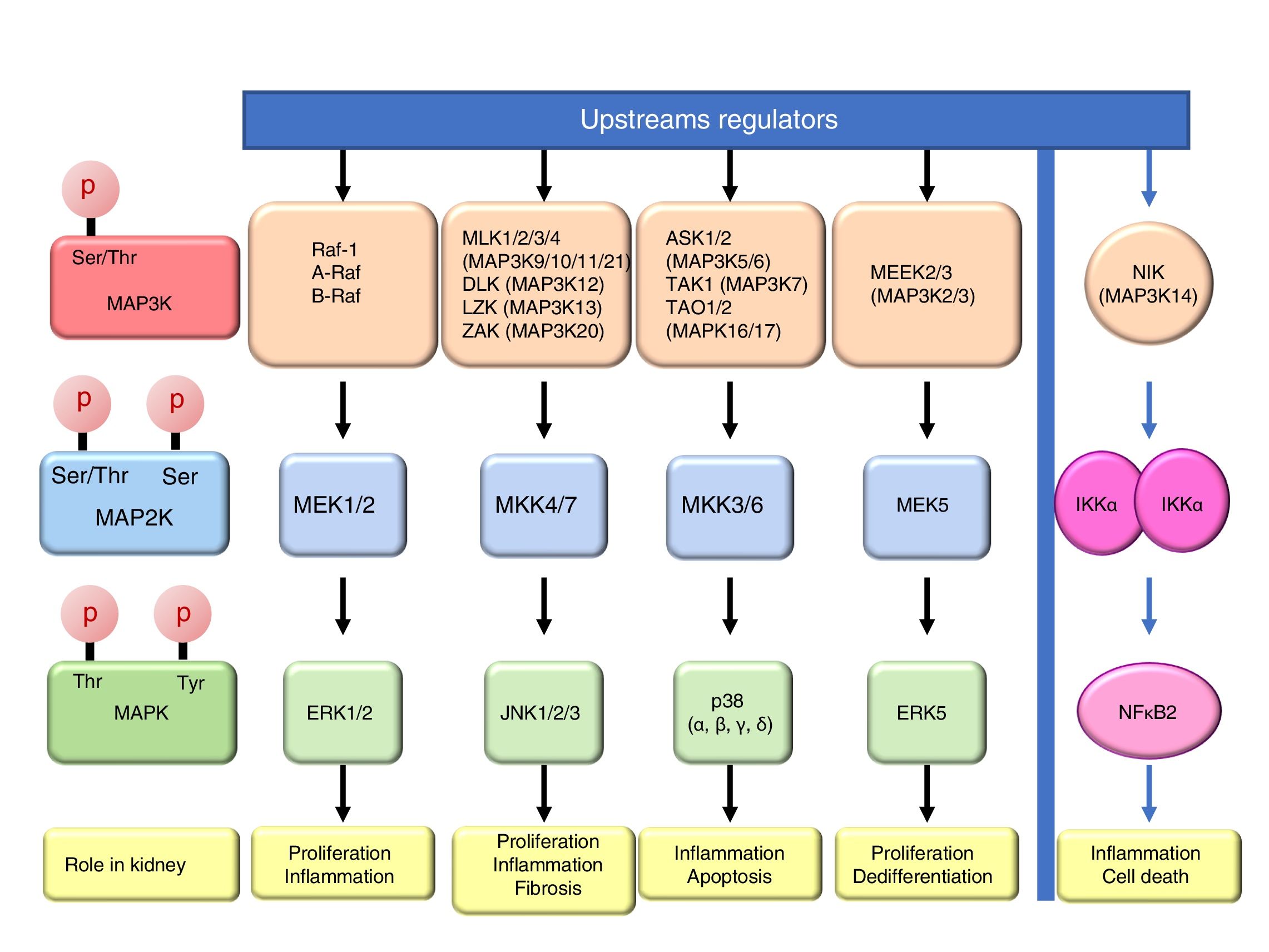
Mitogen-activated protein kinase cascadesMitogen-activated protein kinases (MAP kinases) are functionally connected kinases that regulate key cellular process involved in kidney disease such as all survival, death, differentiation and proliferation. The typical MAP kinase module is composed by a cascade of three kinases: a MAP kinase kinase kinase (MAP3K) that phosphorylates and activates a MAP kinase kinase (MAP2K) which in turn phosphorylates a MAP kinase (MAPK) (Fig. 1). MAP3Ks are activated by MAP4Ks, oxidative stress, small proteins G or by TRAF adaptor proteins in response to growth factors, inflammatory cytokines, drugs, or physical stress. There are 24 characterized MAP3K, named from MAP3K1 to MAP3K21 plus RAF1, BRAF, and ARAF. Most of them are expressed by normal kidneys (Fig. 2).11
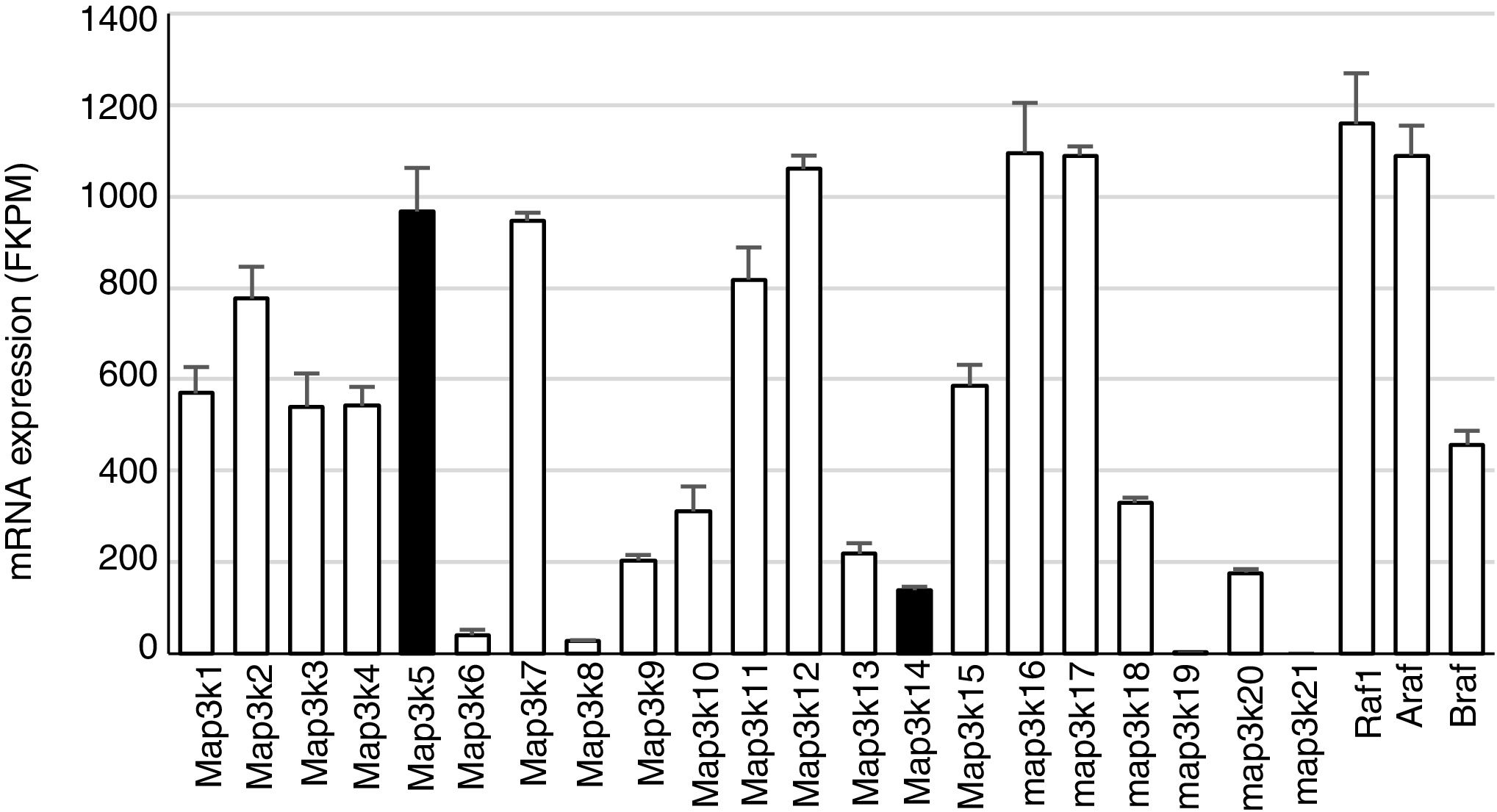
Constitutive MAP3K expression in murine kidneys. Data obtained by RNA-seq analysis of three normal murine kidneys. Mean±SD of gene expression in kidneys from 3 wild-type control mice.11
The final targets of MAPK modules are transcription factors or other kinases that regulate gene expression by transcriptional or post-transcriptional mechanisms.12 There are four well known and conventional MAPK pathways: the ERK pathway, the JNK pathway, the p38 pathway and the ERK5 pathway.13,14 The ERK pathway was the first characterized MAPK cascade. It is formed by three MAP3Ks RAF1, ARAF and BRAF, two MAP2Ks, MEK1 and MEK2 and two MAPK, ERK1 and ERK2, collectively known as ERK1/2. The ERK pathway has multiple functions, and is generally associated to survival and proliferation in different tissues including kidney.15,16 However, additional functions have been described. As examples, in the kidney ERK pathway mediated TWEAK-induced proliferation and pro-inflammatory response in tubular cells.15,17 The JNK pathway is composed by seven MAP3K, MLK1–4 (respectively MAP3K9–11 and MAP3k21), DLK (MAP3K12), LZK (MAP3K13) and ZAK (MAP3K20); two MAP2Ks (MKK4/7) and three MAPKs (JNK1, JNK2, JNK3). It is generally related to cell proliferation, and it has also been linked to the development of renal fibrosis and inflammation in cyclosporine nephrotoxicity.18–21 The p38 pathway is formed by 5 MAP3Ks, ASK1/2 (MAP3K5/6 respectively), TAK1 (MAP3K7), TAO1 (MAPK16), and TAO2 (MAP3K17); two MAP2Ks MKK3/6 and four MAPKs p38 (α, β, γ, δ). This pathway regulates and frequently promotes renal inflammation and apoptosis.22,23 As examples, p38 mediates proliferation of tubular cells induced by TWEAK and expression of inflammatory mediators in renal cells induced by MIF.15,24 The ERK5 pathway is composed by 2 MAP3K, MEEK2/3 (MAP3K2 and 3 respectively); MAP2K MEK5 and MAPK ERK5. ERK5 is the only ERK protein that contains a nuclear localization signal it appears to regulate cell proliferation and dedifferentiation in the kidney, like ERK1/2.18,25,26
ERKs 3, 4, 7 are atypical pathways and the upstream MAPKs involved in their activation and function are poorly characterized, and some evidences indicate that could be activated by autophosphorylation.18 Moreover, ERK 3 and 4 could also be activated by Group I p21-activated Kinases (PAKs) indicating that they are not activated by a typical MAPK module.27 ERK8 is the most recently identified protein kinase that is regulated by autophosphorylation and specific upstream activating kinase are unknown.28
Additional MAP3K are not associated with specific MAPK cascades. One example is NF-kappa-B-inducing kinase (NIK), initially identified as a kinase that participates in NFκB activation. The amino-acid sequence reveled that it is a serine/threonine kinase like MAP3Ks, although the signaling pathway is not a classical MAPKs module.29 NIK is a key protein for non-canonical, NFκB2 activation and is required for the physiological immune response and also promotes inflammation. In the kidney, NIK targeting may be protective during kidney injury.10,30
Drugs targeting MAP3K kinasesA search in Scifinder (https://scifinder.cas.org) for small molecule inhibitors of MAP3K, made by searching individual molecules by their official gene symbol and alternative names, disclosed that small molecule inhibitors were under study for 14 of them (Table 1). Inhibitors for five MAP3K are in clinical use for malignancy, although in some cases MAP3K inhibition is a promiscuous effect of a drug targeted to a different kinase.
MAP3K inhibitors. List according to a search in Scifinder (https://scifinder.cas.org) by their official gene symbol and alternative names performed on September 15, 2018. The same table with references can be found as l.
| Gene symbol (name) | Inhibitors | Representative inhibitor | Inhibitors in clinical use |
|---|---|---|---|
| MAP3K1 (MEKK1) | No | No | |
| MAP3K2 | Yes | No | |
| MAP3K3 (MEKK3) | No | No | |
| MAP3K4 | No | No | |
| MAP3K5 (ASK1) | Yes | GS-4997 (Phase 2) | No |
| MAP3K6 (ASK2) | No | No | |
| MAP3K7 (MEKK7, TAK1) | Yes | 5Z-7-oxozeaenol | No |
| MAP3K8 (TPL2) | Yes | No | |
| MAP3K9 | No | No | |
| MAP3K10 | No | No | |
| MAP3K11 (MEKK11, MLK3) | Yes | URMC-099 | No |
| – | |||
| – | |||
| MAP3K12 (MUK, DLK) | Yes | – | Sunitinib (Sutent, SU11248) |
| CEP-1347 | |||
| GNE-3511 | |||
| GNE-8505 | |||
| MAP3K13 (LZK) | Yes | – | No |
| MAP3K14 (NIK) | Yes | – | No |
| NIK SMI1 | |||
| Patent | |||
| Patent | |||
| MAP3K15 (ASK3) | No | – | No |
| MAP3K16 (TAO1, TAOK1) | Yes | No | |
| Patent | |||
| MAP3K17 (TAO2, TAOK2) | Yes | Patent | No |
| – | |||
| RAF1 (CRAF) | Yes | Regorafenib (Stivarga)Inhibition of RAF-1>BRAF | |
| Sorafenib (Nexavar; Sorafenibum; BAY 43-9006) Inhibition of RAF1>BRAF | |||
| Vemurafenib (Zelboraf, PLX4032/RG7204/RO5185426), inhibition of RAF1 ∼BRAF | |||
| GW5074 | Dabrafenib(Tafinlar, GSK2118436) inhibition BRAF>RAF1 | ||
| PLX-4720 | Lifirafenib (BGB-283)Inhibition RAF1>BRAF | ||
| AZ628 | |||
| ZM 336372 | |||
| ZM 336372 | |||
| GW 5074 | |||
| RAF265 | |||
| NVP-BHG712 | |||
| TAK-632 | |||
| RAF709 | |||
| CCT196969 | |||
| CCT241161 | |||
| BAW2881 (NVP-BAW2881) | |||
| LY3009120 | |||
| BRAF | Yes | SB-590885/SB-699393 | Encorafenib (Braftovi, LGX818) selectivity for BRAF |
| AZ304 | |||
| GDC-0879 | |||
| CEP-32496 | |||
| RO5126766 (CH5126766) | |||
| ARAF | Yes | LY3009120 | Lifirafenib (BGB-283)InhibitionA-RAF>RAF1>BRAF |
| ZAK (MLT, MLTK, HCCS-4, MRK, AZK) | Yes | Sorafenib | |
| PLX-4720 | Vemurafenib | ||
| AST-487 | Imatinib (Gleevec) | ||
| Nilotinib (Tasigna) | |||
| Ponatinib (Iclusig) | |||
| Motesanib/AMG-706 | |||
| Bofutinib/SKI-606 (Bosulif) |
Table 2 summarizes current knowledge about MAP3K involvement in non-malignant kidney disease. Our group has been instrumental in developing the evidence base supporting the involvement of MAP3K14/NIK as a pathogenic molecule in kidney disease. However, the potential role of most MAP3K in kidney disease remains unexplored in vivo.
MAP3K and non-malignant kidney injury.
| Gene symbol (name) | Expression/activation in kidney injury | Targeting in kidney injury | Ref. |
|---|---|---|---|
| MAP3K1 (Mekk1) | Protective? Mekk1 directly represses PKD1 transcription in cultured cells. No in vivo studies | [112] | |
| MAP3K5 (ASK1) | Increased: activity in glomerular and/or tubular epithelial cells in IRI-AKI, DKD, glomerulopathies (crescentic glomerulonephritis/experimental membranous nephropathy) and kidney fibrosis induced by UUO | Protective. Targeting Ask1 with inhibitors and/or genetic deficiency protective in AKI, glomerular injury (crescentic glomerulonephritis), diabetic kidney disease and kidney fibrosis (UUO). Clinical trials in DKD did not meet endpoints. | [63–65,68,70,71,113] |
| MAP3K7 (MEKK7, TAK1) | Increased: TAK1 increased in cisplatin-induced AKI, but not in UUO in mice. | Protective: Conditional TAK1−/− protected from kidney fibrosis (UUO); TAK1 inhibition protected from cisplatin-induced or IRI AKI and IRI fibrosis. | [77–79] |
| Deleterious: MAP3K7−/− spontaneous neonatal kidney scarring with retention of embryonic nephrogenic rests. | [80] | ||
| MAP3K8 (TPL2) | Cot/Tpl2 phosphorylated in IRI | Protective: Cot/Tpl2−/− mice protected from IRI: decreased apoptosis | [81] |
| MAP3K14 | MAP3K14 upregulation in folate-induced AKI | Protective: MAP3K14 KO protected from folate and cisplatin-induced AKI | [10] |
| MAP3K15 (ASK3) | Deleterious: hypertensive phenotype of ASK3−/− mice | [82] | |
| BRAF (v-RAF) | Deleterious: BRAF inhibitor nephrotoxicity in humans: AKI and podocyte injury | [104,106] |
AKI: acute kidney injury; IRI: ischemia/reperfusion injury; DKD: diabetic kidney disease; UUO: unilateral ureteral obstruction
MAP3K14/NIK has been conclusively demonstrated to be activated in experimental kidney disease and to contribute to the severity of experimental kidney injury (Fig. 3). NIK is the essential upstream serine/threonine kinase of the non-canonical NFκB pathway.29 It is now known that NIK protein levels are regulated both transcriptionally (as in AKI and in tubular cells in response to TWEAK) and post-transcriptionally.10 Thus, concentrations are low in quiescent cells because of low mRNA levels and rapid degradation of the protein. Cytokines and oxidative stress may increase NIK mRNA as well as protein stability, leading to NIK activation.10,31 NIK induces IκB kinase-α (IKK-α)-mediated phosphorylation of NFκB2 p100, and phosphorylated p100 undergoes ubiquitination and subsequent proteasomal processing to active NFκB2 p52.32 Efficient NFκB2 p100 ubiquitination requires Uba3 (ube1c) and Ube2m (UBC12), ube2d3 (UBCH5c) and intact Cullin1 in SCFβ−TrCP.33 Interestingly, the Ube2m E2 ubiquitin conjugating enzyme and cullin-1 of the SCFβ−TrCP E3 ubiquitin ligase were upregulated in AKI.10 Additional functions of NIK have been recently reported, but their role in kidney injury remains unclear.34–37
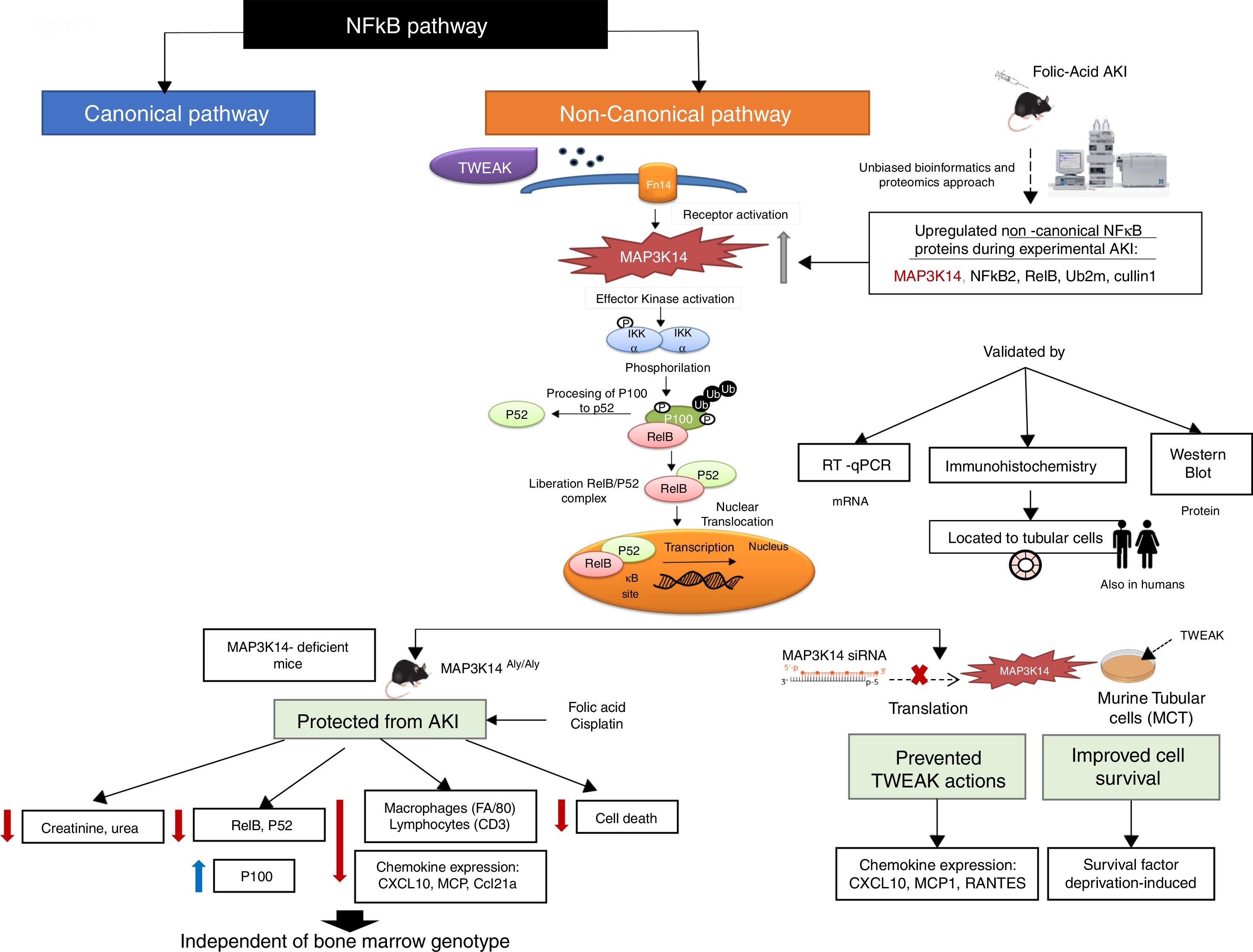
NFκB promotes inflammation in the kidney, as well as having multiple other functions such as promoting cell survival or decreasing the expression of anti-inflammatory proteins such as Klotho and PGC1α in response to TWEAK or tissue injury, while the involvement of NFκB in other TWEAK-dependent responses such as induction of kidney-protective CCl20 expression has not been characterized.38–44 Recent studies have focused on NFκB activity modulation by proteins such as Bcl3 and NFκBiz.45,46 Canonical NFκB activation, a rapid process triggered by multiple stimuli has been best characterized and is responsible for most activities of TWEAK in cultured tubular cells, including expression of most chemokines47 although some (e.g. CCL21) were early on show to be dependent on non-canonical NFκB activation.30 Although non-canonical NFκB activation has been reported in several kidney disease, the pathogenic role of NIK has been best characterized in experimental AKI induced by folic acid (crystal nephropathy) or cisplatin.30,48,49 Evidence for NIK activation in vivo in AKI included increased NIK mRNA and protein levels, NFκB p100 processing to NFκB p52, increased nuclear localization and DNA binding activity of p52/RelB and decreased kidney inflammation, cell death and fibrosis and preserved renal function in NIK activity deficient mice, which were protected from AKI (Fig. 2).10 Upregulation of tubular cells NIK was observed in both human AKI and CKD, suggesting that NIK maybe a therapeutic target in the clinical settings. Protection from AKI may depend on systemic NIK deficiency (e.g. leukocyte NIK deficiency) and/or kidney NIK deficiency. Cell culture and bone marrow transplantation experiments support the hypothesis that renal cell NIK targeting is necessary for nephroprotection. NIK and non-canonical NFκB gene targets have been characterized in the immune system, but there was little prior information on kidney cells.38 In this regard, NIK siRNA targeting reduced inflammatory responses and serum deprivation-induced death in cultured tubular cells.10 Interestingly, NIK targets in tubular cells included both known non-canonical (e.g. CCl21, CXCL10) and canonical (MCP1, Rantes) NFκB pathway targets.10,30 These results were consistent with decreased inflammation and tubular cell apoptosis in vivo in NIK activity-deficient mice during AKI. These results are also consistent with observations targeting another component of the non-canonical NFκB pathway, RelB. Thus, RelB targeting by siRNA protected mice against lethal kidney ischemia-reperfusion, and RelB targeting in cultured proximal tubular cells, protected from apoptosis induced by TNF-cisplatin.50,51
Since NIK deficiency results in immune deficiency a potential role of immune cell NIK in kidney injury cannot be completely excluded. Thus, NIK inhibitors will be expected to target both renal cells and leukocytes and may have adverse effects related to immune suppression. The successful use of NIK inhibitors in experimental systemic lupus erythematosus, and, specifically, for lupus nephritis, should be viewed in the context of their immunosuppressive properties.52 Thus, systemic lupus erythematosus is an autoimmune kidney disease and the current standard of therapy for lupus nephritis is immune suppression.
Non-canonical NFκB activation also contributes to podocyte injury induced by TWEAK activation of this receptor Fn14.49,53 In cultured podocytes, TWEAK increased the expression of the chemokines CCL21, CCL19 and RANTES. The CCL19 and the early RANTES responses depended on canonical NFκB activation, while the CCL21 response depended on non-canonical NFκB activation and could be inhibited by a specific NIK siRNA. Increased kidney Fn14 and CCL21 expression was also observed in podocytes in rat proteinuric kidney disease induced by puromycin, supporting the in vivo relevance of the findings.49 In this regard, there is experimental and human kidney disease evidence supporting a role of TWEAK/Fn14 in promoting podocyte injury in proteinuric kidney disease.53 The potential contribution of NIK to fibroblast proliferation and activation induced by TWEAK and to kidney fibrosis54,55 or in endothelial cell responses to TWEAK56 has not been explored. In this regard, TWEAK, as other inflammatory cytokines, promotes vascular calcification and there is evidence of the cooperation between canonical and non-canonical NFκB signaling, since a siRNA targeting RelB reduced by 20% TWEAK pro-calcific effects and decreased TWEAK-induced loss of human vascular smooth muscle cells contractile phenotype and MMP9 activity, while other effects were prevented by agents blocking canonical activation.57,58
The p52/RelB heterodimers characteristic of NIK-initiated non-canonical NFκB activation share a number of gene targets with RelA-containing, classically activated NFκB complexes.59–61 Since canonical NFκB activation is an early transient response peaking at around 1–3h while and non-canonical NFκB activation is delayed and peaks at around 24h, non-canonical NFκB activation may contribute to sustained NFκB-dependent gene expression.38,62
ASK1/MAP3K5Apoptosis signal-regulating kinase 1 (ASK1) is activated by oxidative stress and, in turn, activates MAPK p38 and JNK and induces apoptotic, inflammatory, and fibrotic signaling in settings of oxidative stress.23 There is convincing evidence of the pathogenic role of ASK1 in experimental kidney disease derived from the study of specific ASK1 inhibitors or ASK1 deficient mice and a clinical trial focused on diabetic kidney disease (DKD) (Table 2).
The first reports focused on AKI. Ask1−/− mice are healthy and are protected from AKI induced by ischemia–reperfusion injury (IRI), while Ask1−/− cultured tubular cells are protected from hypoxia-induced cell death and transfection with dominant-active ASK1 induced apoptosis.63,64 Additionally, transfection of tubular cells with dominant-active ASK1 induced apoptosis decreased chemokine production. Evidence from Ask1 involvement in AKI is also derived from studies with small molecule inhibitors. NQDI-1, an ASK1 inhibitor attenuated renal dysfunction and histological changes in rats with IRI-AKI.65 In the same model, Oral GS-444217 reduced serum creatinine and urea, tubular cell death and proinflammatory and profibrotic mediators in the kidney.23 GS-444217 is a selective ATP-competitive small-molecule inhibitor of ASK1 that inhibits ASK1 autophosphorylation and ASK1-mediated signaling in cells and in vivo.23 Nephroprotection may expand to additional kidney diseases characterized by tubular injury, such as myeloma kidney, which is triggered by the presence in urine of massive amounts of immunoglobulin free light chains. Thus, in cultured human proximal tubular cells, free immunoglobulin light chains increased Ask1 phosphorylation and Ask1 siRNA targeting protected tubular cells from light chain-induced apoptosis.66
Protection by Ask1 targeting has also been reported for experimental glomerular injury. In glomerular epithelial cells (GEC), sublytic complement activation induced oxidative stress and activated Ask1, JNK and p38 MAPK and this was decreased by a dominant negative ASK1 mutant. ASK1 promoted complement-mediated lysis and this was dependent on p38 MAPK activity.67 The in vivo relevance of these findings was supported by the observation of increased glomerular ASK1 activity during experimental membranous nephropathy, a proteinuric kidney disease characterized by podocyte (glomerular epithelial cell).67 However, in vivo targeting was not attempted. More recent reports explored Ask1 inhibitors. In accordance to prior observations of MAPK p38 and JNK activation and the beneficial effect of their targeting in experimental crescentic glomerulonephritis, GS-444217 decreased glomerular inflammation and fibrosis, proteinuria and preserved renal function in diverse rat experimental models of crescentic glomerulonephritis.68
Unilateral ureteral obstruction (UUO) is used as a model of accelerated CKD that recapitulates most of the human features of the disease.69 p38 and JNK were activated in kidney fibrosis induced by UUO. In Ask1−/− mice with ureteral obstruction, p38 was not activated, JNK activation was decreased and kidney inflammation and fibrosis were milder. The key cell targets responsible for kidney protection appeared to be the tubular cells, since tubular cell p38 activation was observed in vivo and Ask1-deficient cultured tubular cells were protected from angiotensin II and H2O2, induced p38 activation and upregulation of TGFβ1, and chemokines, while Ask1−/− fibroblast activity was not impaired.70
However, the studies with more translational potential explored DKD. The ASK1 pathway is activated in glomeruli and tubules of human and experimental DKD. GS-444217 reduced inflammation and fibrosis, prevented albuminuria development and preserved renal function in experimental DKD and the effect was observed on top of renin–angiotensin system blockade.23 However, in earlier studies in streptozotocin-induced diabetes in hypertensive endothelial nitric oxide synthase (Nos3)-deficient mice, GS-444217 did not decrease hypertension or albuminuria, but decreased diabetic glomerulosclerosis and inflammation and reduced renal function.71 A Phase 2 placebo-controlled clinical trial investigating the effects of three different doses of selonsertib (GS-4997) on eGFR (primary end-point) and albuminuria (secondary endpoint) at week 48 in patients with type 2 diabetes was completed in 2016.72 The fact that in a phase 2 RCT the primary endpoint was ambitious, impact on eGFR and not albuminuria is unusual73 and likely reflects the lack of impact of Ask1 inhibition in at least some experimental models of DKD.71 While the results of the clinical trials have not been reported in full, Gilead Sciences announced in 2016 that selonsertib did not meet endpoints in DKD or pulmonary arterial hypertension, but clinical development continues with active phase 3 trials for nonalcoholic Steatohepatitis (NASH: NCT03053063, NCT03053050) that completed enrollment in 2018.74 Although as of December 6, 2018 there is no planned or ongoing selonsertib trial in DKD in clinicaltrials.gov, clinical development for kidney disease appears to continue since phase 1 RCT were registered in the Australia New Zealand Clinical Trial Registry (ANZCTR) and other countries for patients with CKD in the summer of 2018.75 Furthermore, at the 2018 American Society of Nephrology Renal Week, a post hoc analysis of the phase 2 DKD trial was presented.76 The analysis excluded 2 sites for technical deviations as well as patients with baseline eGFR <20mL/min/1.73m2. These exclusions may represent the identification of issues that blurred the information provided by the trial and considered not promising in 2016. In this post hoc analysis, the highest dose of selonsertib (18mg) was associated with significantly milder eGFR slopes than placebo both in the overall trial population and in a high risk population defined by high baseline soluble TNFR1 (sTNFR1) levels.76
Other MAP3K kinases: MAP3K7, MAP3K8 and MAP3K15Additional MAP3K have been targeted in vivo in experimental kidney disease (Table 2). MAP3K7 (MEKK7, TAK1) was increased in cisplatin-induced AKI, but not in UUO in mice.77–79 However, for kinases activity is more relevant that levels of expression. In this regard, conditional TAK1−/− mice were protected from kidney fibrosis (UUO) and TAK1 inhibition with small molecules protected from cisplatin-induced or IRI AKI and IRI fibrosis.77–79 Specifically, in UUO, TAK1 deletion resulted in suppression of JNK, p38, and NF-κB activation, and milder inflammation and fibrosis.77 Despite this success, caution should be exercised in the extrapolation of the results to the clinic, since MAP3K7 appears to have a key role in kidney development and developmental genes may be re-expressed during kidney disease and contribute to kidney repair. Thus, MAP3K7−/− mice develop spontaneous neonatal kidney scarring with retention of embryonic nephrogenic rests.80
There is also some experience targeting other MAP3K in vivo. MAP3K8 (TPL2/Cot) is phosphorylated during IRI and Cot/Tpl2−/− mice were protected from IRI and apoptosis was decreased.81 By contrast MAP3K15 (ASK3) may have a role in protecting from hypertension. Thus, ASK3−/− mice have a hypertensive phenotype.82
Lessons from kinase inhibitors in clinical useCurrent available treatments for CKD are based on renal angiotensin-system (RAS) blockade and immunosuppressive drugs and can delay the development of renal failure but inflammation and fibrosis keep progressing.73,83,84 Several orally available kinase inhibitors, are currently in the pipeline for the kidney disease.63 Data obtained from the clinical use of kinase inhibitors outside nephrology may help to define the role of target kinases in the human context, be it either because of the development of adverse effects or, as described for the Nrf2 activator bardoxolone, because kidney function or adverse events are observed to improve.83,85 In any case, nephrotoxicity observed with the high doses used to treat cancer does not preclude a potential therapeutic effect of a lower dose in kidney disease, although it does provide a note of caution.
Since the approval of Imatinib, in 2001, more than 30 small-molecule kinase inhibitors are being used or tested in cancer treatment86–89 and inflammatory diseases.90,91 Most of the approved kinase inhibitors are used to treat malignancy.87,92–94 or, as is the case for JAK inhibitors baricitinib and tofacitinib, autoimmune disease.95,96 Baricitinib also reduced albuminuria in patients with DKD.73,97 Some kinase inhibitors are nephrotoxic. The most consistent nephrotoxicity is associated with drugs targeting the VEGF pathway and its tyrosine kinase receptor VEGFR, which can induce hypertension, albuminuria, nephrotic syndrome or even thrombotic microangiopathy, and this is consistent with preclinical advances on its role in endothelial and podocyte well-being.98,99 Some kinase inhibitors are promiscuous and, thus it is difficult to related side effects with inhibition of a specific kinase. This is the case for sorafenib, which inhibits VFGFR, PDGFR and BRAF, among others.
There are fewer drugs in clinical use targeting MAP3K. The ones available may inhibit several MAP3K, and thus, it is difficult to pinpoint the exact kinase responsible for any kidney actions. However, nephrotoxicity has been reported for drugs that among other targets, inhibit BRAF (Table 1). Vemurafenib and dabrafenib are BRAF inhibitors approved for treatment of late-stage melanoma.100 Vemurafenib has been associated to the development of AKI. Decreased of renal function (mean reduction of 29ml/min) was associated with nephrotic range proteinuria in 5 patients.101,102 A subsequent study in 74 patients treated with vemurafenib, found severe but reversible AKI predominantly in men with proven tubular and interstitial damage in renal biopsy within the first 3 months of treatment.103 In the FDA review on Adverse Event Reporting Systems of Vemurafenib, there were 132 cases of AKI, 85 in men, and 47 in women.104 A recent report observed AKI in 25% of melanoma patients treated with a combination therapy of the MEK inhibitor cobimetinib and vemurafenib, which was 60% lower than in prior results with vemurafenib monotherapy.105 Dabrafenib also has nephrotoxic potential, with 13 cases of AKI detected form April 2013 to June 2014, mostly in men (12 men vs 1 women).104 Podocyte injury leading to nephrotic developed in one patient during dabrafenib and trametinib treatment.106 In cultured podocytes, BRAF inhibition decreased PLCɛ1 and nephrin expression, inhibited the podocyte VEGF system and increased the permeability to albumin.106 A patient on encorafenib and binimetinib, BRAF and MEK inhibitor, respectively, developed after two months renal dysfunction and proteinuria due to crescentic glomerulonephritis with a granulomatous reaction which reversed following withdrawal of the chemotherapy.107 In any case, although clinical nephrotoxicity is observed when used for malignancy, it is still worth testing these agents for experimental kidney disease in case a lower dose may favorable impact kidney injury. We should remember that the first reports on captopril and the kidney referred to AKI and glomerular injury at a time when considerably higher doses were used.108–111
ConclusionsMAP3K sit at the cusp of intracellular signaling cascades, making them potential targets for therapeutic intervention. However, for the very same reason they may impact multiple cell processes. They may be targeted by small molecules, but the similarity between them is hindrance to the development of specific inhibitors. Most of them remain unexplored from the kidney point of view and this is an opportunity. Human experience is for the most part limited to small molecules used in cancer therapy. In this regard, glomerular and tubular nephrotoxicity has been described, especially for BRAF inhibitors. However, there is experimental evidence derived from genetically modified mice and/or the in vivo use of small molecule inhibitors for a pathogenic role of MAP3K14/NIK, ASK1/MAP3K5, MAP3K7/MEKK7/TAK1, and MAP3K8/TPL2/Cot in AKI and/or CKD. Even a clinical trial of ASK1/MAP3K5 targeting was performed in DKD, although the results have not been published. Given the safety up to now of ASK1/MAP3K5 inhibitors in humans and the wide range of responsive experimental nephropathies to MAP3K14/NIK, ASK1/MAP3K5, MAP3K7/MEKK7/TAK1, and MAP3K8/TPL2/Cot, further research should be aimed at defining the optimal drug, dose and disease target for human drug development as well as to define if there is any pathogenic role in kidney disease for those MAP3K still unexplored in the kidney.
Conflict of interestThe authors declare no conflict of interest.
Grant support: Instituto de Salud Carlos III (ISCIII) and FEDER – Fonds Européen de Développement Économique et Régional (FEDER) funds EUTOX, CP14/00133, PI15/00298, PI16/01900, PI16/02057, PI18/01366, Sociedad Española de Nefrologia, Fundacion Renal Iñigo Alvarez de Toledo (FRIAT), ISCIII Red de Investigacion Renal (REDinREN) RD016/009, Salary support: ISCIII Miguel Servet to MDSN, Universidad Autónoma de Madrid to LC.
The following is the supplementary data to this article:




