









The major contributions of Fuller Albright to our understanding of calcium and phosphorus regulation and primary hyperparathyroidism are highlighted. Albright was the first investigator to initiate a systematic study of mineral metabolism. With resources limited to the measurement of serum calcium and phosphorus and the infusion of parathyroid extract, Albright used balance studies to establish a framework for our understanding of calcium and phosphorus regulation and primary hyperparathyroidism. Albright was the first to show that the etiology of primary hyperparathyroidism could be from either an adenoma or hyperplasia of the parathyroid glands and stone disease was a separate manifestation of primary hyperparathyroidism. Albright also showed that: 1) a renal threshold for calcium excretion was present in hypoparathyroid patients; 2) correction of hypocalcemia in hypoparathyroid patients with vitamin D had a phosphaturic action; 3) renal failure reduced the intestinal absorption of calcium in primary hyperparathyroidism; 4) the «hungry bone» syndrome developed after parathyroidectomy in severe primary hyperparathyroidism; and 5) a target organ can fail to respond to a hormone. He also suggested that a malignant tumor could be responsible for ectopic hormone production. Finally, our review integrates the observations of Albright with our current knowledge of calcium regulation and disorders.
INTRODUCTION
Fuller Albright’s academic career began in the late 1920s and ended in 1956 after brain surgery for Parkinson’s disease resulted in a non-functional state until his death in 1969. Although increasingly disabled by Parkinson’s disease from the mid 1930s, Albright continued to make important contributions to our knowledge of calcium and phosphorus disorders1. His book, "Parathyroid Glands and Metabolic Bone Disease", published in 1948, is a testimony to his many important observations2. Our goal is to highlight some of the many contributions made by Albright on calcium and phosphorus regulation and primary hyperparathyroidism and to integrate the findings of Albright with more recent studies. Albright’s contributions to our understanding of renal phosphate transport have been discussed elsewhere3. Virtually all that Albright observed remains valid today, but as often happens, the explanations and their complexity continue to evolve.
Our retrospective highlights Albright’s enduring legacy to the modern study of calcium and phosphorus regulation and primary hyperparathyroidism. The following topics are discussed: 1) the resources available to Albright to study calcium and phosphorus regulation and primary hyperparathyroidism; 2) Albright’s studies of calcium balance; 3) phosphaturia resulting from the correction of hypocalcemia in hypoparathyroid patients; 4) primary hyperparathyroidism as a surgically curable disorder; and 5) several clinical vignettes which include: a) a prelude to the trade-off hypothesis of Slatopolsky and Bricker advanced by Albright to explain the development of secondary hyperparathyroidism in renal failure; b) the suggestion that a malignant tumor could be responsible for ectopic hormone production; c) the realization that vitamin D deficiency can be associated with the failure to respond to parathyroid hormone (PTH); and d) the appreciation that immobilization can be a cause of hypercalcemia.
TOOLS OF THE TRADE
In the late 1920s when Albright first started his studies of calcium and phosphorus regulation and primary hyperparathyroidism, his primary tools were: 1) parathyroid extract (PTE), which was a bovine preparation from Eli Lilly and Company; 2) the personnel on the newly organized Ward 4 at Massachusetts General Hospital who were trained to perform balance studies in which dietary calcium and phosphorus could be prepared with accuracy and the fecal and urine excretion of calcium and phosphorus collected with precision4; and 3) the measurement of calcium and phosphorus in the blood, urine and feces. As a result of balance studies it became possible to determine how variations in dietary calcium and phosphate content and the administration of PTE affected: 1) serum calcium and phosphorus concentration; 2) fecal and urinary excretion of calcium and phosphorus; and 3) retention or loss of calcium and phosphorus from the body (figure 1). Because PTH could not be measured, Albright could not know whether PTH values were modified by serum calcium.
EFFECT OF PARATHYROID EXTRACT AND DIETARY CALCIUM ANS PHOSPHATE ON CALCIUM BALANCE IN NORMAL SUBJECTS AND IN PATIENTS WITH HYPOPARATHYROIDISM AND HYPERPARATHYROIDISM
In an early study, Albright infused PTE into a patient with longstanding idiopathic hypoparathyroidism (Figure 2)5. Albright observed that: 1) urine phosphorus excretion immediately increased and peaked within two hours; 2) the increase in serum calcium and decrease in serum phosphorus followed the increase in phosphorus excretion; and 3) there was a critical serum calcium value at about 8.5 mg/dl at which a negligible urinary calcium excretion suddenly became appreciable. Albright concluded that the action of PTE was rapid, its first effect was phosphaturia, and the increase in serum calcium followed the increase in phosphorus excretion. Thus, Albright developed the hypothesis that PTH primarily modified phosphorus rather than calcium, a view that he still championed when his book was published almost 20 years later2. Albright argued that the PTH-induced increase in phosphorus excretion and the resulting decrease in serum phosphorus promoted bone dissolution releasing calcium. While such a conclusion might sound somewhat fanciful today, it should be remembered that Albright had participated in studies in which ammonium chloride-induced acidosis increased the serum calcium concentration and urinary calcium excretion without increasing intestinal calcium absorption suggesting that acidosis induced bone dissolution6,7. Today it is recognized that phosphate depletion increases calcium release from bone resulting in hypercalciuria and even hypercalcemia despite a marked reduction in PTH values suggesting that bone dissolution is an important feature8,9. Finally, Albright was correct in his hypothesis that phosphate was a major modifier of the calcemic response of bone to PTH. Many subsequent studies have shown that phosphate loading and restriction change the calcemic response to PTH10-12.
As already mentioned, Albright had observed during a PTE-induced increase in serum calcium in a hypoparathyroid patient that the serum calcium threshold at which urinary calcium excretion increased from negligible values was approximately 8.5 mg/dl. However, that threshold value did not account for the now known effect of PTH on urinary calcium excretion. In 1961, one of the authors (CRK) first showed that PTE administration directly increased renal calcium reabsorption13. Subsequently, it was shown that the threshold serum calcium value for excreting calcium during a calcium infusion was seen at serum calcium values between 7 and 8 mg/dl in hypoparathyroid patients14. In 1984, Ogata and associates showed that besides PTH, the active form of vitamin D, calcitriol, directly increased the threshold for renal calcium excretion and also enhanced the responsiveness of the tubule to PTH15. More recently, Bindels and colleagues have shown that the stimulatory effect of both calcitriol and PTH on renal calcium reabsorption results from the activation of an epithelial calcium channel (TRPV5) in the distal convoluted tubule16.
VITAMIN D TREATMENT AND CALCIUM INFUSION AS PHOSPHATURIC AGENTS
In 1938 and in 1942, Albright used the newly available analog of vitamin D, dihydrotachysterol, for the treatment of hypocalcemia in patients with hypoparathyroidism17 and also in the newly described disorder of pseudohypoparathyroidism in which there was a failure to respond to administered PTE18. Albright observed that the correction of hypocalcemia increased urine phosphate excretion. This observation had been made earlier by associates of Albright19 and by Howland and Kramer20. In 1965, Eisenberg demonstrated in hypoparathyroid patients that the phosphaturia was independent of vitamin D by showing that a prolonged intravenous infusion of calcium sufficient to normalize the serum calcium concentration at 48 hours was phosphaturic and also lowered the serum phosphorus concentration (figure 3A, figure 3B)21. Evidence has accumulated during the past several years that the recently discovered bone-derived phosphaturic hormone, fibroblast growth factor 23 (FGF23) might be involved. High dietary calcium has been shown to stimulate FGF2322 and the correlation between the serum calcium concentration and FGF23 seen in primary hyperparathyroidism23-25 even remained significant after parathyroidectomy24. However, a study has yet to be performed identifying the specific mechanism for the observation made more than 70 years ago by Albright and others.
PRIMARY HYPERPARATHYROIDISM, INTESTINAL CALCIUM ABSORPTION, AND RENAL FAILURE
In patients with primary hyperparathyroidism, Albright showed that changes in dietary calcium and phosphate affected calcium balance. The first patient studied was Captain Martell26, who was to have seven parathyroid operations before an ectopic parathyroid gland was removed from the anterior mediastinum27. From the balance studies in Captain Martell, who had serum calcium values between 13.1 and 15.3 mg/dl, Albright made the following observations: 1) the patient’s negative calcium balance on a low calcium diet (0.1 grams/day) was greater than in normal controls (-0.46 vs -1.29 grams per 3 day study period); 2) the increased negative calcium balance in the hyperparathyroid patient was due to increased urinary calcium excretion because fecal calcium excretion was less than in normals; 3) an infusion of PTE in normal volunteers sufficient to increase serum calcium to between 11.5 and 12.8 mg/dl resulted in a negative calcium balance duplicating the results of the hyperparathyroid patient; and 4) a calcium diet of 1.07 grams/day in the hyperparathyroid patient resulted in a positive calcium balance (0.36 grams per day) because urinary calcium excretion increased by only 0.06 grams/day from that on a low calcium diet (0.1 grams/day). Thus, Albright concluded that in primary hyperparathyroidism: 1) adaptation to a low calcium diet did not occur because high urine calcium losses persisted; 2) intestinal calcium absorption was increased; and 3) high PTH values were the likely cause of the negative calcium balance.
Albright also evaluated the effect of dietary phosphate on calcium balance in hyperparathyroidism. He showed that a high phosphate diet improved calcium balance in hyperparathyroid patients on both low (0.07 g/day) and normal (0.9 g/day) intakes of dietary calcium28. When high dietary phosphate was given to patients with primary hyperparathyroidism, there was: 1) almost complete intestinal absorption of phosphate; 2) rapid excretion of the absorbed phosphate by the kidneys; 3) a rise in the previously low serum phosphorus; 4) a fall in the previously elevated serum calcium; 5) a rise in the serum calcium-phosphorus product; and 6) a fall in urinary calcium excretion. While a high phosphate diet seemed to have certain beneficial effects such as lowering the serum calcium concentration and decreasing urinary calcium excretion, Albright recognized that there were two potential dangers associated with increased phosphate ingestion in patients with primary hyperparathyroidism: 1) parathyroid poisoning with soft tissue calcium deposition including the kidney and lungs; and 2) calcium-phosphate kidney stones. Today, the recognition that hyperphosphatemia in CKD patients and perhaps even high normal serum phosphorus values in the general population are associated with increased vascular disease and mortality probably from increased vascular calcification29,30 could be considered an extension of the pioneering studies of Albright.
By 1934, Albright came to understand that renal failure had a specific effect on calcium and phosphorus regulation31. When a 13 year old girl with moderate renal failure was referred for hypercalcemia (13.6 mg/dl), bone demineralization, a markedly elevated serum alkaline phosphatase, and a serum phosphorus of 4.3 mg/dl, balance studies were performed before the parathyroid surgery during which an adenoma was removed. The balance studies showed that on a low calcium diet (0.1 g/day), urine calcium excretion despite hypercalcemia was only marginally greater than in controls on a similar diet (94 mg vs 63 mg/day). This value was much less than in non-azotemic patients with primary hyperparathyroidism and hypercalcemia (435 mg/day). Also, fecal calcium excretion was greater than in controls and much greater than in hyperparathyroid patients without renal failure. Because of the results in this young girl with renal failure, Albright reviewed the results of the series of balance studies which had been performed on Captain Martell before and after he developed renal failure. As shown in table 1, for a similar magnitude of hypercalcemia (14 mg/dl vs 14.4 mg/dl), for the same low calcium diet after the onset of renal failure, intestinal calcium absorption was less (0.06 vs 0.26 g/day) as was urine calcium excretion (0.09 vs 0.44 g/day). Thus, Albright was the first to recognize that the development of renal failure in primary hyperparathyroidism dramatically decreased both intestinal absorption and the renal excretion of calcium. Today, it is known that PTH stimulates renal production of the active form of vitamin D, calcitriol, which in turn enhances intestinal absorption of calcium. With loss of renal function, calcitriol production is decreased despite high PTH values, a result which may in part be due to increased FGF23 values. Consequently, both intestinal calcium absorption and renal calcium excretion are reduced in renal failure. The latter results from both a decrease in the glomerular filtration of calcium and increased tubular calcium reabsorption from high PTH values.
PRIMARY HYPERPARATHYROIDISM
In several patients in his original series of 17 patients published in 193432, Albright made the diagnosis of hyperparathyroidism only because he had the insight to measure serum calcium and phosphorus values in all patients who presented with kidney stones. In the first 14 patients to undergo parathyroid surgery, parathyroid adenomas were found. Two of these patients had ectopic parathyroid adenomas located in the anterior mediastinum. Cases 15 to 17 had hyperplasia of all the parathyroid glands. Because hyperplasia had not been previously recognized as an entity, Case 15 required three parathyroid operations to remove a sufficient amount of the hyperplastic glands before the hypercalcemia resolved. Between the second and third operations, estrogen treatment and irradiation of the pituitary and parathyroid glands were tried without success33.
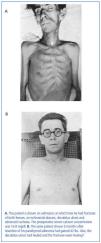
In this series of 17 patients, the dimensions, but not the weights of the removed parathyroid glands were provided32. The mean amount of parathyroid tissue removed per patient was approximately 83 times greater than the combined size of four normal human parathyroid glands, which subsequently were shown to have a combined weight of approximately 140 mg34. Based on these results, the average estimated weight of removed parathyroid tissue for each patient was approximately 11 grams. In severe cases of primary hyperparathyroidism with marked hypercalcemia, cachexia and debilitating fractures were sometimes seen and parathyroidectomy was life saving (figure 4). The different magnitude of primary hyperparathyroidism in patients presenting in the 1930s and today is shown by the very high preoperative serum calcium values in Albright’s patients (figure 5). Finally, Albright was the first to describe the "hungry bone syndrome" in which severe hypocalcemia developed values shortly after parathyroidectomy (figure 6). He also showed that the decrease in serum calcium correlated with the pre-operative serum alkaline phosphatase value. When the serum calcium decreased to less than 7 mg/dl, Albright reported that tetany and visual disturbances were often seen. In actuality, the subsequent recognition of the "hungry bone syndrome" in dialysis patients after parathyroidectomy is an extension of the results in primary hyperparathyroidism by Albright.
In 1934, Albright also recognized that patients with primary hyperparathyroidism presented with either bone disease or stone disease, but rarely both together32. In patients with bone disease, skeletal symptoms associated with bone loss, bone cysts, brown tumors, and fractures predominated. In patients with stone disease, presenting symptoms were those associated with nephrolithiasis and skeletal problems were generally absent. Albright questioned why there should be two separate presentations for the same disease. He hypothesized that the extent of bone disease was proportional to the duration of disease times the daily loss of calcium32. Thus, according to Albright a short duration of disease would lessen the risk of bone disease. Moreover, a high calcium intake would make bone disease less likely because as Albright had previously observed in studies of patients with primary hyperparathyroidism, a high calcium diet resulted in a positive calcium balance26.
In 1945, Keating reported 24 patients with primary hyperparathyroidism in whom the magnitude of hypercalcemia, hypophosphatemia and serum alkaline phosphatase elevation was greater in patients with bone disease than with stone disease35. In contradiction to the hypothesis advanced by Albright, the patients with bone disease had a shorter duration of symptoms before presentation. In 1956, Dent also reported that patients with bone disease had a shorter duration of symptoms36. Subsequently, Dent reported that there was no difference in dietary intake of calcium between patients with bone or stone disease37.
In 1968, Lloyd reviewed 138 consecutive cases of primary hyperparathyroidism accumulated by Dent in London from 1950 to 196538. Patients were divided into overt bone disease without kidney stones (Type 1, n = 44) and kidney stones without overt bone disease (Type 2, n = 88). The remaining six patients had neither overt bone disease nor kidney stones. As shown in table 2, the adenoma weight was greater, the growth rate of the parathyroid tissue more rapid, and the duration of symptoms was shorter in patients with bone disease. These results contradicted Albright’s hypothesis that patients with bone disease have a longer duration of disease.
One explanation for the shorter duration of disease together with more severe hypercalcemia and larger adenomas in patients with bone disease is simply a more rapid growth rate of the parathyroid adenomas in patients with bone disease. In the early 1970s, both Woodhouse et al39 and Lumb and Stanbury40 suggested that the more rapid growth of adenomas in patients with bone disease might be from a lack of vitamin D. In 1987, one of the authors (CRK) wrote an editorial in support of this possibility41. Also in 1987, Paillard and associates reported that patients with bone disease had lower values of the stored form of vitamin D, 25-hydroxyvitamin D (25[OH]D), 3.4 ± 2.0 vs 17.6 ± 13.6 ng/ml (P <0.001) and three-fold greater PTH values than patients with stone disease42. In more recent studies, Silverberg et al in a longitudinal study of patients with mild hyperparathyroidism, found that patients in the lowest tertile of 25(OH)D measurements (12 ± 3 ng/ml) had the highest PTH values43. Rao, et al., in a study of 148 consecutive patients operated for primary hyperparathyroidism with a mean resected parathyroid gland weight of 1.27 grams, reported an inverse correlation between 25(OH)D and parathyroid gland weight while the correlation between the active form of vitamin D, calcitriol, and parathyroid gland weight was not significant44. These results suggest that a suboptimal vitamin D status may stimulate parathyroid adenoma growth. The presence of 1 alpha-hydroxylase, the enzyme responsible for conversion of 25(OH)D to calcitriol, in parathyroid cells suggests the possibility that 25(OH)D may directly affect PTH secretion and parathyroid gland growth45,46. Also contributing to the lowering of 25(OH)D levels in primary hyperparathyroidism is that high PTH levels decrease 25(OH)D levels by increasing conversion of 25(OH)D to calcitriol47. In summary, even in the studies of Silverberg and Rao in which the increased weight of the parathyroid adenoma was modest and the diagnosis of primary hyperparathyroidism was made relatively early in the course of the disease, vitamin D status seemed to play a role43,44.
Since the 1970s, the clear demarcation between patients with bone and stone disease previously seen in patients with primary hyperparathyroidism starting with the report of Albright in 1934 and continuing through the 1960s has been lost32,35,38,48-51. Beginning in the 1930s and continuing for the next three decades, the sequence of events in diagnosing bone disease in primary hyperparathyroidism often was as described by Albright, "appearance of a bone tumor, biopsy, diagnosis of benign giant cell tumor, local treatment, and finally recognition of generalized disease only years later"2. However, the introduction of multichannel analyzers in which serum calcium and phosphorus values were routinely measured resulted in the detection of many asymptomatic hyperparathyroid patients with mild hypercalcemia.
In a recent review of primary hyperparathyroidism, the average weight of the removed adenoma was 400 to 600 mg, values which are only three to four times greater than the combined weight of four normal parathyroid glands52. In contrast, the mean weight of the removed parathyroid adenomas in the Albright study from 1934 was approximately 11 grams32. In 1947, Norris reviewed 322 cases from the existing world literature and reported that the average weight of a removed parathyroid adenoma was 8 grams48. In 1963, Hodgkinson reported that the mean weight of the removed adenoma was 5.1 grams in patients with bone disease and 1.4 grams in patients with stone disease49. Similar differences in the weight of parathyroid adenomas between patients with bone and those with stone disease were reported by Lloyd in his analysis of Dent’s patients38 and by O’Riordan51. The mean weight of the parathyroid adenoma in patients with bone disease was 5.9 grams in the former and 4.2 grams in the latter series.
In areas of the world with limited access to medical care, vitamin D insufficiency/deficiency appears to be more common and the duration of primary hyperparathyroidism much longer before diagnosis and treatment. In recent studies of primary hyperparathyroidism from India and China, the presence of large parathyroid adenomas and bone disease has been associated with vitamin D insufficiency/deficiency (figure 7)53-57. In a study from China, the presenting PTH value was 21 times greater than normal55. Similarly, in a study from India, the mean weight of the removed parathyroid adenoma was 10.75 g in hypercalcemic patients and 3.9 g in normocalcemic patients54. In another study from India, the mean weight of the removed parathyroid adenoma was 7.9 g56. Moreover, the 25(OH)D value was less than 10 ng/ml in the majority of these patients. In the cited studies, pathologic bone fractures, bone cysts, and brown tumors were commonly encountered54-56. In summary, the severe form of primary hyperparathyroidism characterized by large adenomas and disabling bone disease, first described by Albright in the 1930s, is still commonly encountered in areas of the world with limited access to medical care. Furthermore, the severe form of primary hyperparathyroidism seen in these patients is often associated with vitamin D insufficiency/deficiency.
VIGNETTES IN WICH OBSERVATIONS BY ALBRIGHT HAVE HAD CONTINUED CLINICAL RELEVANCE
1937. Prelude to the "Trade-Off Hypothesis" of Slatopolsky and Bricker10,58 which was Advanced to Explain the Development of Secondary Hyperparathyroidism
Albright made the following statement in a 1937 publication59: "It has been suggested above that the parathyroid hyperplasia is a compensation for the disturbed equilibrium occasioned by phosphate retention resulting from the renal insufficiency. In the absence of the hyperplasia there would probably be greater phosphate retention in the blood with a lowering of the blood calcium level and severe tetany. If these concepts are correct, the hyperplasia would have to be considered beneficial. The one reservation might have to be made that, while helping homeostasis, the parathyroid hyperplasia may lead to bone disease". Confirmation of the Albright hypothesis has been shown in many animal and clinical studies of phosphate loading. Also in studies of patients with stage 3 and 4 CKD treated with the calcimimetic, cinacalcet, the reduction in PTH values has increased the serum phosphorus concentration60.
1941. Hypothesis that Hypercalcemia in Malignancy could be from Ectopic Hormone Production
At a clinicopathological conference, a 51 year old male presenting with hypercalcemia and hypophosphatemia was discussed61. A neck exploration for presumed hyperparathyroidism was performed, but no abnormality was found. Bone x-rays showed a destructive lesion in the right ilium, which on biopsy was reported to originate from a renal cell carcinoma. The comment of Albright at the end of the conference was "Why a person should have high serum calcium and low serum phosphorus when the cause of the disturbance is a tumor destroying bone is an interesting theoretical question. We treated this case by radiation of the tumor masses; the serum calcium went down to normal, and the serum phosphorus went up to normal. Gradually, both values became abnormal again. I suspected that the tumor might be producing parathyroid hormone. I therefore had it assayed by Dr. J. B. Collip, but no hormone was found". In essence, Albright was the first to suggest the possibility of ectopic hormone production by a tumor. Today we know the causal agent responsible for the hypercalcemia and hypophosphatemia to be PTH-related protein and not PTH.
1941. Pseudohypoparathyroidism in Vitamin D Deficiency
A year before Albright reported the clinical entity of pseudohypoparathyroidism in which there was a failure to respond to PTE in patients with characteristic body features18, he recognized a subset of patients with vitamin D deficiency in whom the serum calcium was low and the serum phosphorus was normal. Albright logically but incorrectly thought that the problem was because the necessary compensatory increase in parathyroid function had not taken place62. Subsequently, there continued to be reports of patients with vitamin D deficiency in whom hypocalcemia was accompanied by normal or even high serum phosphorus values40,63-65. In 1985, Rao et al established that the hypocalcemia associated with vitamin D depletion can impair the phosphaturic response to PTH despite an appropriately increased nephrogenous cyclic AMP response66. Correction of hypocalcemia with vitamin D and calcium treatment restored the phosphaturic response to PTH despite a reduction in nephrogenous cyclic AMP. Thus, these patients had an acquired form of pseudohypoparathyroidism type II, which was subsequently shown to be from a postreceptor G protein defect67. In the opinion of the authors, another possibility which should be evaluated is that the vitamin D deficiency and the hypocalcemia combine to impair FGF23 production22-25.
1941. Immobilization and Hypercalcemia
Albright reported the case of a 14 year old boy who in an athletic injury, fractured his right femur at the site of a bone cyst68. After an open reduction, the boy was placed in a spica cast immobilizing him from the waist down. Shortly thereafter anorexia and vomiting developed. Because the presence of the bone cyst raised the possibility of primary hyperparathyroidism, serum calcium was measured and found to be 14.6 mg/dl with a serum phosphorus of 4.5 mg/dl. Mild renal insufficiency also developed. Because of the persistent hypercalcemia, the boy underwent a parathyroid exploration at which only normal parathyroid glands were identified. Six weeks later because of persistent hypercalemia, the anterior mediastinum was explored but no parathyroid tissue was identified. Once the patient was mobilized, the serum calcium decreased to normal values. Albright concluded that the sequence of events was: a) solitary bone cyst; b) fracture through cyst; c) immobilization of a large part of a previously active skeleton; d) osteoporosis from disuse with an excess of calcium from bone being presented to the kidney; e) an inability of the kidney to excrete promptly all the calcium; f) resulting in hypercalcemia; and g) kidney damage from excess of calcium being excreted through the kidney. Albright further added that once renal insufficiency developed, it probably increased the tendency to hypercalcemia. From this case, Albright learned that immobilization of an individual with active skeletal remodeling increases calcium efflux from bone and he also recognized that a decreased glomerular filtration rate reduces the capacity to excrete calcium, which in turn, exacerbates hypercalcemia.
CONCLUSION
In the late 1920s, Albright joined Joseph Aub and Walter Bauer to pursue studies of calcium and phosphorus metabolism. First Aub and then Bauer were appointed to academic positions in other subspecialties at Harvard, which by 1930 left the young Albright as the primary investigator of calcium and phosphorus metabolism in Boston. Through inductive reasoning, which has been defined by the late Jacob Bronowski as that unpredictable blend of speculation and insight, Albright came to recognize in normal volunteers and in hypo- and hyperparathyroid patients, the presence of consistent patterns of response for calcium and phosphorus metabolism. Fuller Albright was truly the first person to establish a sense of order out of the existing chaos in the new field of calcium and phosphorus metabolism. As one of the 20th century’s preeminent philosophers of science, Karl Popper has stated, "Science does not rest upon rock-bottom. The bold structure of its theories rises, as it were, above a swamp, but not down to any natural or given base; and when we cease our attempts to drive our piles into a deeper layer, it is not because we have reached firm ground. We simply stop when we are satisfied that they are firm enough to carry the structure, at least for the time being". Albright was the first to establish a functional system which explained calcium and phosphorus metabolism. His classic book, The Parathyroid Glands and Metabolic Bone Disease, published in 1948, became the standard reference for a generation of students of calcium and phosphorus metabolism. Albright trained many future investigators and became an inspiration for the next generation of clinical investigators studying calcium and phosphorus disorders. As the importance of translational research is again rightfully being emphasized, the contributions of Fuller Albright should be recognized and he should be celebrated as a role model for a new generation of young clinical investigators.

Table 1. A comparison of the calcium and phosphorus metabolism of a patient with hyperparathyroidism before and after the development of renal impairment

Table 2. Comparison of two types of primary hyperparathyroidism based on a series analyzed by Lloyd

Figure 1. Graphic representation of balance data.

Figure 2. Effect of parathyroid extract on urinary calcium and phosphorus excretion in a patient with idiopathic hypoparathyroidism.

Figure 3. Effect of a 48 hour calcium infusion with normalization of the serum calcium concentration on phosphate excretion in hypoparathyroid patients.

Figure 4. The same patient before and after parathyroidectomy from original series of Albright.

Figure 5. Average preoperative serum calcium values of the first 35 patients with primary hyperparathyroidism from the Massachusetts General Hospital series.

Figure 6. Fall in serum calcium values after parathyroidectomy in 35 cases of primary hyperparathyroidism (first demonstration of hungry bone syndrome).

Figure 7. A global view of vitamin D levels and the magnitude of hyperparathyroidism as determined by the elevation in PTH.



