
















Haemolytic uraemic syndrome (HUS) is a clinical entity defined as the triad of nonimmune haemolytic anaemia, thrombocytopenia, and acute renal failure, in which the underlying lesions are mediated by systemic thrombotic microangiopathy (TMA). Different causes can induce the TMA process that characterises HUS. In this document we consider atypical HUS (aHUS) a sub-type of HUS in which the TMA phenomena are the consequence of the endotelial damage in the microvasculature of the kidneys and other organs due to a disregulation of the activity of the complement system. In recent years, a variety of aHUs-related mutations have been identified in genes of the complement system, which can explain approximately 60% of the aHUS cases, and a number of mutations and polymorphisms have been functionally characterised. These findings have stablished that aHUS is a consequence of the insufficient regulation of the activation of the complement on cell surfaces, leading to endotelial damage mediated by C5 and the complement terminal pathway. Eculizumab is a monoclonal antibody that inhibits the activation of C5 and blocks the generation of the pro-inflammatory molecule C5a and the formation of the cell membrane attack complex. In prospective studies in patients with aHUS, the use of Eculizumab has shown a fast and sustained interruption of the TMA process and it has been associated with significative long-term improvements in renal function, the interruption of plasma therapy and important reductions in the need of dialysis. According to the existing literature and the accumulated clinical experience, the Spanish aHUS Group published a consensus document with recommendations for the treatment of aHUs (Nefrologia 2013;33[1]:27–45). In the current online version of this document, we update the aetiological classification of TMAs, the pathophysiology of aHUS, its differential diagnosis and its therapeutic management.
El síndrome hemolítico urémico (SHU) es una entidad clínica definida por la tríada anemia hemolítica no inmune, trombocitopenia e insuficiencia renal aguda, en la que las lesiones subyacentes están mediadas por un proceso de microangiopatía trombótica (MAT) sistémico. Distintas causas pueden desencadenar el proceso de MAT que caracteriza el SHU. En este documento consideramos SHU atípico (SHUa) como el subtipo de SHU en el que los fenómenos de MAT son fundamentalmente consecuencia del daño producido en el endotelio de la microvasculatura renal y de otros órganos por desregulación de la actividad del sistema del complemento. En los últimos años se han identificado diversas mutaciones en genes del sistema del complemento asociados a SHUa, que explicarían aproximadamente el 60% de los casos de SHUa, y se han caracterizado funcionalmente numerosas mutaciones y polimorfismos asociados a SHUa que han permitido determinar que la patología se produce como consecuencia de la deficiente regulación de la activación del complemento sobre las superficies celulares y que lleva al daño endotelial mediado por la activación del C5 y de la vía terminal del complemento. Eculizumab es un anticuerpo monoclonal humanizado que inhibe la activación del C5, bloqueando la generación de la molécula proinflamatoria C5a y la formación del complejo de ataque de membrana. En estudios prospectivos en pacientes con SHUa su administración ha demostrado la interrupción rápida y sostenida del proceso de MAT, con una mejora significativa de la función renal a largo plazo y una reducción importante de la necesidad de diálisis y el cese de la terapia plasmática. En función de las evidencias científicas publicadas y la experiencia clínica acumulada, el Grupo Español de SHUa publicamos un documento de consenso con recomendaciones para el tratamiento de la enfermedad (Nefrología 2013;33(1):27–45). En la presente versión online del documento se actualizan los contenidos sobre la clasificación etiológica de las MAT, la fisiopatología del SHUa, su diagnóstico diferencial y su manejo terapéutico.
Haemolytic uraemic syndrome (HUS) is a clinical entity consisting of the triad of nonimmune microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal failure.1 The histological lesions of HUS typically involve systemic thrombotic microangiopathy (TMA), largely resulting in impaired intrarenal vessels. A greater number of HUS cases are caused by a Shiga toxin-producing (STEC) Escherichia coli enteric infection or verotoxin-producing (VTEC) germs, resulting in the so-called typical HUS or STEC (VTEC)-HUS. Genetic or acquired (autoantibodies) dysregulation of the alternative complement pathway leading to endothelial damage and systemic TMA phenomena occur in nearly 10% of HUS reports.2 This kind of HUS related to the complement dysregulation is known as atypical HUS (aHUS).
In 2011, Eculizumab (Soliris®; Alexion Pharmaceuticals, Connecticut, USA) was approved by the American and European regulatory agencies for the treatment of aHUS.3 Eculizumab is a humanised monoclonal antibody inhibiting C5 activation and blocking the production of the proinflammatory C5a anaphylatoxin, as well as the formation of the membrane-attack complex, leading to cell lysis.4 In prospective studies conducted in aHUS patients, Eculizumab effectively halted the TMA process and its effects, and was associated with the rapid, significant, and long-term improvement of haematological and renal function abnormalities,5 and with improved systemic involvement and high blood pressure.
In 2012, the Spanish Group for aHUS gathered to develop a consensus document including recommendations for treating the disease.6 The group has been meeting every year ever since then in order to update both the understanding of the various aspects of interest related to the disease (including the aetiological classification of TMAs, the pathophysiology of aHUS, and companion diagnostics) and treatment recommendations based on already published scientific evidence and clinical experience. The contents originally published in Nephrology 2013;33(1):27–45 have been updated in the current online version of the consensus document.
Aetiological classification of thrombotic microangiopathiesThe term TMA describes a histological lesion of the arterioles and capillaries resulting in thickened and swollen vessel walls, detachment of endothelial cells, widening of the subendothelial space caused by the build-up of proteins and cell lysis material, and the presence of platelet thrombi obstructing vascular lumen (Fig. 1).1 Two clinical entities with different aetiology and pathophysiology are characterised by primary TMA lesions: thrombotic thrombocytopenic purpura (TTP) and HUS.
 Ischaemic and retracted glomeruli. (B) Mesangiolysis (C) Thrombi in the glomerular capillaries (arrow). (D) Artery occluded by platelet thrombi. Photographs courtesy of Dr. R. Ortega (Histopathology department of the Hospital Universitario Reina Sofía, Córdoba).")
Renal histopathological lesions from haemolytic uraemic syndrome. (A) Ischaemic and retracted glomeruli. (B) Mesangiolysis (C) Thrombi in the glomerular capillaries (arrow). (D) Artery occluded by platelet thrombi. Photographs courtesy of Dr. R. Ortega (Histopathology department of the Hospital Universitario Reina Sofía, Córdoba).
Intravascular thrombosis in TTP results from a severe deficiency in the metalloprotease activity of the A Desintegrin and Metalloproteinase with ThromboSpondin type 1 motif, member 13 (ADAMTS13), a plasma enzyme responsible for splitting the ultra-large multimers of the Von Willebrand factor.7 This deficiency may be genetic or acquired via IgG circulating antibodies blocking ADAMTS13 (particularly in patients receiving platelet antiaggregants).8
Ninety percent of HUS cases are caused by a STEC enteric infection resulting from contaminated food (typical HUS or STEC [VTEC]-HUS).2 The Shiga toxin causes a direct injury on the vascular endothelium, triggering a number of cell and vascular events which ultimately lead to TMA.2 Clinical presentation usually involves abdominal pain and diarrhoea, together with acute renal failure within 4–10 days. Prognosis is typically favourable, with a mortality rate below 5% and 80% of patients achieving complete clinical recovery, although progression to severe chronic renal failure is observed over time in up to 20–30% of patients.9,10
aHUS is essentially diagnosed by exclusion once ADAMTS13 (TTP) deficiency or STEC infection (STEC-HUS) are ruled out. In patients with aHUS, TMA phenomena are a consequence of the dysregulation of the alternative complement pathway on the cell surface. This abnormality results in uncontrolled activity on own cells following complement activation (by several triggering factors), leading to endothelial damage, inflammation, and secondary thrombosis, with an increasing number of cases involving genetic or acquired factors. Mutations have been described in one or more complement proteins in nearly 60% of over 1000 patients with reports of aHUS in the literature,11–18 although a genetic component (involving complement genes or other, including coagulation genes) and/or unspecified autoimmunity may also be present in the remaining patients. Of note, anti-Factor H (FH) autoantibodies have been found in 5–10% of aHUS patients.19 Unlike STEC-HUS, which is usually an isolated event, aHUS is a chronic and relapsing entity triggered by the uncontrolled activation of the complement system. Before Eculizumab became available, aHUS had been mostly associated with a poor prognosis: the mortality rate following a first episode of aHUS was 10–15%, and renal function remained unrecovered in up to 50% of patients.11,12,20
A type of aHUS resulting from recessive mutations in the DGKE gene coding for the DGK-¿ (diacylglycerol kinase-¿) protein has recently been described.21 The loss of this enzyme activity in endothelial cells, platelets, and podocytes leads to endothelial cells apoptosis and impaired angiogenic response, thereby resulting in a prothrombotic and inflammatory state.22 Patients with DGKE mutations exhibit various phenotypes ranging from aHUS to membranoproliferative glomerulonephritis with high proteinuria and nephrotic syndrome.23 aHUS patients develop persistent high blood pressure and haematuria-proteinuria (including in the nephrotic range) in their first year of life. Unlike paediatric aHUS associated with complement genetic alterations, progression to chronic renal disease among these patients is not sudden, but develops over years.21
In addition to STEC enteric infection (typical HUS), abnormalities in the regulation of complement activation, mutations in DGKE or coagulation genes (aHUS), or (genetic or autoimmune) deficiency of ADAMTS13 in TTP, there are many other factors and clinical entities that may be associated with the development of TMA. This kind of TMA is included under the term secondary TMA. Some cases reported in children are associated with methylmalonic aciduria24 or more commonly (5% of HUS reports in children) with neuraminidase-producing invasive Streptococcus pneumoniae infections (resulting in the exposure of the crypto-antigen T in the cell surface and unleashing the TMA phenomenon),25 or H1N1 infection.26 TMA has been generally associated with viral infections (CMV, HIV, parvovirus), neoplastic processes, drugs (antitumor agents, including the vascular endothelial growth factor inhibitors, immunosuppressants such as calcineurin inhibitors [cyclosporine and tacrolimus] or the mammalian target of rapamycin inhibitors [mTOR; sirolimus, everolimus], platelet antiaggregants, antivirals, or oral contraceptive drugs), malignant high blood pressure, bone marrow or solid organ transplantation, pregnancy and postpartum, autoimmune systemic diseases, or glomerulonephritis.27
Importantly, the foregoing causes of TMA may not always be identified in all patients, whereas some may present more than one aetiology, resulting in a heterogeneous presentation and a challenging diagnosis. In fact, overlapping entities are common and up to 25% of patients with STEC-HUS and 86% of patients with pregnancy-associated HUS develop complement system mutations, where aHUS is actually the underlying disease.28,29 Mutations in the complement system have also been reported in post-transplant HUS associated with the use of calcineurin inhibitors and in HUS related to autoimmune diseases in 27% and 33% of patients, respectively.12 Furthermore, several cases of secondary TMA have been reported to date with successful treatment outcomes with Eculizumab (TMA associated with drugs,30 solid organs31 or bone marrow32 transplantation, pregnancy33 and systemic erythematous lupus34). The fact that complement blockade (by Eculizumab) is associated with a favourable clinical response and the reversibility of TMA suggests the potential and important role of non-genetic complement dysregulation in many cases of secondary TMA, predisposing patients to its development. On this basis, Fig. 2 summarises the proposed aetiological classification of TMAs and illustrates the potential overlapping between these clinical entities. The classification of TMAs should be understood as a current topic of interest and major debate is taking place among the medical community as a result of the continuous progress made in the understanding of the pathophysiology of these entities.35 Given that the aHUS mediated by complement dysregulation is the main reason for debate, only this entity will be discussed in the following sections.

Classification of the aetiologies of thrombotic microangiopathies. ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; aHUS: atypical haemolytic uraemic syndrome; CMV: cytomegalovirus; FB: complement factor B; FH: complement factor H; FI: complement factor I; HCV: hepatitis C virus; HELLP: Hemolysis, Elevated Liver enzymes, Low Platelet count; HIV: human immunodeficiency virus; HUS: haemolytic uraemic syndrome; MCP: membrane cofactor protein; mTOR: mammalian target of Rapamycin; SEL: systemic erythematous lupus; STEC: Shiga toxin-producing Escherichia coli; THBD: thrombomodulin; TMA: thrombotic microangiopathy; TTP: thrombocytopenic thrombotic purpura; VEGF: vascular endothelial growth factor.
aHUS is considered an ultra-rare disease. Data available on its incidence and prevalence are limited, as well as the knowledge of the actual epidemiology of the disease. The annual incidence of aHUS in the US has been estimated to be of ∼1–2cases/million inhabitants.36 A recent multicentre study in Europe reported an incidence of 0.11cases/million inhabitants. According to the European Medicines Agency (EMA), the prevalence may be of ∼3.3patients/million inhabitants/year among patients below the age of 18, with lower rates among adults.
Children and adults are predominantly affected by aHUS, although it may develop at any time in life.11,12 The onset of the disease usually occurs before the age of 18 (60% vs. 40%) and sex characteristics are well-balanced (women are primarily affected when the disease is developed in adulthood).11,13
Clinical presentationClinical onset is often abrupt, although 20% of patients may develop it progressively (in weeks or months), accompanied by subclinical anaemia, fluctuating thrombocytopenia, and preserved renal function.11 The clinical picture comprises the triad of nonimmune microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal failure.1 High levels of lactate dehydrogenase (LDH), undetectable haptoglobin and schistocytes confirm the presence of intravascular hemolysis20 associated with haematuria, proteinuria, and/or acute renal failure (with or without oligoanuria). High blood pressure resulting from volume overload or vascular lesion is common.1 In some patients, the single manifestation of TMA may be proteinuria with high blood pressure and progressive renal failure without haematological abnormalities.
Even though aHUS lesions are predominantly observed in renal vessels, the diffuse and systemic nature of the TMA phenomenon leads to the involvement of the microvasculature of other organs (including, but not limited to the brain, heart, intestine, pancreas, and lungs),1 therefore accounting for common extrarenal symptoms.11,12 Neurological symptoms are the most common (48%),37 including irritability, somnolence, confusion, convulsions, encephalopathy, stroke, hemiparesis, visual abnormalities, hemiplegia, and coma.1,12,37,38 Myocardial infarction has been described in up to 3% of aHUS patients in relation to sudden death.12,39 Myocardiopathy, heart failure, and peripheral ischaemic vasculopathy have also been reported,19,37,40,41 as well as diarrhoea (30%) and other digestive symptoms (including, but not limited to colitis, nausea, vomit, abdominal pain, hepatitis, cholestasis, and pancreatitis).12,19,38,42 Skin involvement including ulcer lesions in lower limbs has recently been reported in aHUS patients.43 Heterogeneity of symptoms has posed a challenge for companion diagnostics of other causes of TMA.
PathophysiologyThe complement system, consisting of several circulating plasma and membrane-associated proteins, is part of innate immunity and is vital for fighting infections, processing immune complexes, antibody response, and the elimination of apoptotic residues. Activation by any of the existing pathways (classical, lentin, and alternative) leads to the formation of multiprotein complexes with C3-convertase activity splitting the C3 protein and resulting in C3b (Fig. 3). The covalent binding of this molecule to the surfaces activating the complement favours phagocytosis by polymorphonuclear leukocytes and macrophages, therefore resulting in activated C5, directing the attack complex to the membrane and causing cell lysis. In addition, the resulting C3b leads to a rapidly enhanced complement activation by promoting the formation of further C3-convertases, since it is one of the components of C3-convertase of the alternative pathway.44 In order to avoid total uptake by complement activation, as well as damage to self tissues (C3b binds indiscriminately to both pathogens and self cells), a number of process-regulating proteins, such as FH, the membrane cofactor protein (MCP), and complement factor I (FI) dissociate C3-convertases and result in C3b degradation. C3b levels, therefore, remain low under normal conditions and they build up following complement activation only in the structures related to this activity.
 leads to the build-up of large quantities of C3b on the activator cell membrane, causing opsonisation and C5 activation (terminal or lytic pathway), resulting in the formation of the membrane attack complex and cell lysis. Complement activation results in inflammation and leucocyte recruitment. The key process in complement activation is C3b formation, which depends on unstable enzymatic complexes – C3-convertases – catalysing the rupture of C3 to create C3b. In turn, C3b has the ability to form further C3-convertase of the alternative pathway (C3bBb), thus enhancing the initial activation. The mediation of C3B production is two-fold: dissociation of C3-convertases and proteolytic inactivation of C3b and C4b. Several regulatory proteins in plasma and the cell membrane carry out this regulatory activities, including, factor H, MCP and factor I, which play an essential role in the dissociation of C3-convertase of the alternative pathway (C3bBb) and the proteolytic degradation of C3b. Mutations in these proteins found in patients with aHUS interfere with this regulatory activity of the alternative pathway activation. Some patients with aHUS are carriers of mutations in proteins C3 and factor B organising C3-convertase. These mutations are particular, as they increase the activity of mutated proteins (gain-of-function mutations), resulting in increased complement activation and exceeding the capacity of regulatory proteins.")
Complement dysregulation in atypical haemolytic uraemic syndrome. Complement activation by any of the 3 pathways (detection of foreign antigens, alternative pathway; of antibodies, classical; or mannan polysaccharides, lectin) leads to the build-up of large quantities of C3b on the activator cell membrane, causing opsonisation and C5 activation (terminal or lytic pathway), resulting in the formation of the membrane attack complex and cell lysis. Complement activation results in inflammation and leucocyte recruitment. The key process in complement activation is C3b formation, which depends on unstable enzymatic complexes – C3-convertases – catalysing the rupture of C3 to create C3b. In turn, C3b has the ability to form further C3-convertase of the alternative pathway (C3bBb), thus enhancing the initial activation. The mediation of C3B production is two-fold: dissociation of C3-convertases and proteolytic inactivation of C3b and C4b. Several regulatory proteins in plasma and the cell membrane carry out this regulatory activities, including, factor H, MCP and factor I, which play an essential role in the dissociation of C3-convertase of the alternative pathway (C3bBb) and the proteolytic degradation of C3b. Mutations in these proteins found in patients with aHUS interfere with this regulatory activity of the alternative pathway activation. Some patients with aHUS are carriers of mutations in proteins C3 and factor B organising C3-convertase. These mutations are particular, as they increase the activity of mutated proteins (gain-of-function mutations), resulting in increased complement activation and exceeding the capacity of regulatory proteins.
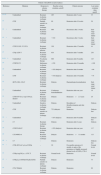
Several studies have shown that around 60% of aHUS patients are carriers of mutations in complement-regulating genes (CFH, MCP, CFI, thrombomodulin [THBD], or in the components of C3-convertase, factor B [FB], and C3).45–54 All these mutations cause the dysregulation of the alternative pathway (Table 1). FH acts in plasma by controlling complement homeostasis and in cell surfaces by preventing damage to self components. Mutations in the C-terminal region of FH are characteristic of aHUS. Because the altered FH region mediates complement activation in cell surfaces, cell protection against accidental damage resulting from complement activation is decreased by these mutations, with no involvement of complement regulation in plasma.55 The functional assay of aHUS-associated mutations found in other complement genes, including MCP, CFI, CFB or C3, has also confirmed that all of them result in a defective protection of cell surfaces and this loss of complement regulation may be due to the decreased activity of regulating proteins or to the abnormally high activity of C3-convertases. Thus, while the regulatory activity of these proteins is impaired by mutations in FH, MCP, and FI, mutations in FB or C3 result in further activation of the C3-convertase.
Risk factors in atypical uraemic haemolytic syndromea
| Mutations |
| Loss of function |
| CFH (∼13%) |
| MCP (∼11%) |
| CFI (∼10%) |
| THBD (∼4%) |
| Gain of function |
| C3 (∼4%) |
| CFB (∼3%) |
| Polymorphisms |
| Increasing risk |
| CFH: c.-332C>T; c.2016A>G (p.Gln672Gln); c.2808G>T (p.Glu936Asp) |
| MCP: c.-652A>G; c.-366A>G; c.989-78G>A; *897T>C |
| Providing protection |
| CFH: c.184G>A (p.Val62Ile) |
| Autoantibodies |
| Anti-FH (∼5%) |
| Environmental factors |
| Infections |
| Immunosuppressants |
| Oral contraceptives |
| Anti-cancer drugs |
Anti-FH: anti-complement factor H antibody; CFB: complement factor B gene; CFH: complement factor H gene; CFI: complement factor I gene; MCP: membrane cofactor protein. Gene; THBD: thrombomodulin gene.
“Multiple hits” theory. aHUS is a complex disease normally involving various risk, genetic, and environmental factors. Patients are commonly carriers of more than one mutation in complement genes or combined mutations with risk polymorphisms. Environmental factors are also necessary to help reveal the genetic disposition from mutations or polymorphisms. Concomitant mutations with risk polymorphisms, autoantibodies, or triggering environmental factors account for the incomplete penetrance of aHUS, as well as for the differences in its presentation and progression among carriers of complement gene mutations.
Around 5–10% of aHUS patients develop anti-FH antibodies targeted to the C-terminal region, with similar effects to those observed in FH mutations.56,57 Their role in the pathogenesis of aHUS has not been fully established, but seems to be associated with disease onset or recurrence. Given that antibody titres may decrease over time, they should be screened early in the course of aHUS. Anti-FH antibodies are associated with complement factor H-related protein 1 deficiency (FHR1) in patients with aHUS.58
Penetrance of aHUS in carriers of mutations in some genes is around 50%, with only a few carriers from families with identified mutations commonly developing aHUS and showing a variable clinical presentation. Clinical heterogeneity, which results from the existence of additional (genetic and environmental) risk factors mediating the development and the outcome of the disease, is largely observed among unrelated carriers of this mutation. Screening for complement mutations in aHUS patients and conducting case-control studies based on genetic polymorphisms in candidate genes or genetic markers in the human genome has allowed for the identification of some variants (polymorphisms) in CFH and MCP genes modulating the penetrance and severity of the disease (Table 1).49,59,60
Haplotypes CFH-H3 and MCPggaac are the most relevant polymorphisms associated with the risk of aHUS. Both haplotypes include single-nucleotide polymorphisms (SNP) in the CFH and MCP gene-promoter region, downregulating FH and MCP. The presence of both polymorphisms in homozygosis may provide a rationale for aHUS disposition in patients with no mutations in any of the genes associated with aHUS. A recent collaborative study by the European Working Party on Complement Genetics in Renal Diseases including 795 patients with aHUS has shown that 3% of these patients were carriers of combined mutations in more than one gene. Additionally, this large study has proved that concomitant risk haplotypes CFH-H3 and MCPggaac also lead to significantly increased disease penetrance in carriers of combined mutations, stressing the idea that genotyping of these risk polymorphisms helps predict the risk of aHUS in mutation carriers.61
Along with the previous genetic alterations, a number of triggering environmental factors are also implicated in the onset of aHUS. The above mutations are predisposing factors of the disease, preventing adequate complement regulation in cell surfaces when the system becomes activated in microvessels. aHUS is triggered by infectious events in 50–80% of patients,11,12,40 particularly those involving the upper respiratory tract (influenza H1N1 virus). Diarrhoea caused by gastroenteritis may precede aHUS in up to 30% of cases (including diarrhoea by STEC11,12,19).12 Pregnancy, particularly during post-partum, is a common predisposing factor of aHUS among women,12,29 together with the use of oral anovulatory agents.
Mutations in the gene coding for thrombomodulin (THBD), an anticoagulant protein acting as thrombin cofactor and also regulating the FI-mediated C3b inactivation, have been associated with aHUS.62 Based on complement dysregulation typical of patients with aHUS, the functional analysis of THBD mutations associated with aHUS has shown that thrombomodulin mutations impair the complement regulatory activity.62 Nevertheless, the impairment of the anticoagulation activity by thrombodulin mutations associated with aHUS and the relevance of these abnormalities in aHUS remains unknown. In this regard, a recent study conducted in 36 patients with aHUS has assessed the presence of mutations in the genes of the complement system and coagulation through massive DNA sequencing, detecting mutations in genes from both systems.63 The gene in the coagulation system with the largest number of mutations was plasminogen (PLG), a cymogene which plays an important role in fibrinolysis following conversion to plasmin. Even though these data suggest that coagulation genes may add to aHUS disposition (particularly PLG), further studies are required to confirm these observations.
The search of new genes associated with aHUS has also been addressed by Lemaire et al.21 through exome sequencing. The authors have identified homozygotic mutations in the DGKE gene coding for the DGK-¿ protein in 13 patients with aHUS from 9 families. These patients had a very early onset of aHUS, generally within their first year of life, followed by multiple recurrences and common progression to end-stage renal failure in the second decade of life.21 Deficiency of DGK-¿ in endothelial cells has been recently shown to induce the expression of ICAM-1 and tissue factor by means of increased p36-MAPK-mediated signalling, leading to apoptosis, altering the angiogenic response, and determining a proinflammatory and prothrombotic phenotype. Yet, the absence of DGK-¿ is not detrimental to complement activation in cell surfaces.21,22 The absence of DGK-¿ in podocytes and endothelial cells may probably impair the diaphragm of glomerular filtration, which would account for massive proteinuria and the susceptibility to glomerular conditions among these patients,21,23 although the reason why these patients tend to develop several glomerular conditions remains uncertain. Finally, despite the role of the complement in the development of renal disease among carriers of DGKE, mutations had been initially ruled out,21 patients with DGKE mutations additionally associated with other genes previously related to aHUS, including THBD and C3,64 have been recently identified, thus suggesting that complement dysregulation may play a role in the modulation of disease onset and outcome at least in some carriers of DGKE mutations.
PrognosisThe availability of Eculizumab has significantly revolutionised the prognosis of patients living with aHUS, a very severe disease in most cases in spite of intensive treatment with plasma therapy (PT; Table 2). Following a first episode of aHUS, overall mortality was higher than 10% and more than half of the patients required dialysis and/or developed a more permanent renal damage in the next 12 months.11,12,20 The clinical outcome changes relatedly depending on the patient's mutation. In this respect, outcome seemed to be particularly poor in patients with FH and C3 mutations, with mortality and end-stage chronic renal failure (ESCRF) rates over 50% within one year from the first episode of aHUS. Furthermore, half of these patients relapsed. Mutations in FI, FB, and THBD were also associated with high rates of mortality and ESCRF at one year (50%), with relapses occurring in nearly one in 3 patients overcoming the first episode of aHUS. On the other side, less than 10% of patients with MCP mutations died or progressed to ESCRF, although the risk of relapse among these patients was higher and up to 90% of them developed new episodes of aHUS. Between 50 and 75% of patients with mutations in FH, CI, C3, FB or THBD died or developed ESCRF within 3–5 years from the first episode of aHUS.1
Clinical outcome of patients with atypical haemolytic uraemic syndrome based on complement abnormalities (prior to Eculizumab).
| Gene | Risk of death or ESRF in the first episode or within the next year | Risk of relapse | Risk of death or ESRF at 3–5 years | Risk of relapse following renal transplant |
|---|---|---|---|---|
| CFH | 50–70% | 50% | 75% | 75–90% |
| CFI | 50% | 10–30% | 50–60% | 45–80% |
| MCP | 0–6% | 70–90% | 6–38%a | <20% |
| C3 | 60% | 50% | 75% | 40–70% |
| CFB | 50% | 3/3 without ESRF | 75% | 100% |
| THBD | 50% | 30% | 54%a | 1 patient |
| Anti-FH | 30–40% | 40–60% | 35–60%a | Higher with increased antibody titres |
Anti-FH: anti-complement factor H antibodies; CFB: complement factor B gene; CFH: complement factor H gene; CFI: complement factor I gene; ESRF: end-stage renal failure; MCP: membrane cofactor protein gene; THBD: thrombomodulin gene.
The outcome of renal transplantation (RT) among patients with ESCRF due to aHUS has been historically limited by the increased percentage of recurrences of post-transplant disease (∼50%; graft loss rate: 80–90%65,66), although it changes significantly based on the type of alteration. In a series of 57 patients with aHUS receiving RT, 5-year recurrence-free graft survival was significantly lower in patients with mutations in the genes coding for complement proteins compared to patients in whom only polymorphisms but no genetic abnormalities were found.67 Yet, it should be stressed that the risk of recurrence of aHUS following RT in patients with no genetic abnormalities is also deemed high. 68 Mutations in FH are associated with a higher risk of recurrence or graft loss following RT (75–90%; specifically those related to abnormalities in terminal 3′ and gene conversion between CFH and CFHR1, resulting in the hybrid gene CFH/CFHR1 [both abnormalities impair the functionality of the C-terminal domain of FH]), posing a high risk with mutations in C3 and FI as well (40–80%; Table 2).12,42,48,65,67,69–71 To date, very few transplants have been performed in patients with FB mutations, though recurrence of aHUS and graft loss were reported in all cases.49,72 In general, plasma factors of the complement involved in aHUS are synthesised in the liver, and so patients with mutations in the complement genes coding for these factors remain prone to aHUS following RT, as dysfunctional factors are still being produced. MCP is a transmembrane protein that is highly expressed in the kidney and, as a result, this defect can be corrected by RT by delivering unchanged MCP into the graft. Over 80% of patients with MCP mutations develop no recurrence of aHUS following RT, with a long-term survival rate comparable to that of patients receiving transplants for other reasons.40,65,66 The risk of post-transplant recurrence in patients with THBD62 mutations or anti-FH antibodies is not well-established, although it seems to be related to high and persistent titres of antibodies in the latter.19,73
Diagnosis of atypical haemolytic uraemic syndromeIn light of the rapid evolution and the severity of TMA, a differential diagnosis should be immediately established from the syndrome perspective, allowing for supportive measures to be taken within 24–48h from patient's admission. Considerations for an aetiological diagnosis of TMA will subsequently be made. Table 3 summarises the major procedures and diagnostic tests recommended for the diagnosis of TMA, including specific tests for companion diagnostics of the various aetiologies of TMA.
Diagnostic tests and procedures recommended for patients with thrombotic microangiopathy.
| General diagnostic tests |
|---|
| • Complete medical records, including drugs, data from systemic diseases, personal and family history• Complete physical examination, including a fundoscopic exam• General routine blood and urine tests• Haptoglobin levels• Serum complement levels• Peripheral blood smear• Serology for systemic diseases (ANA, anti-ADN, ANCA, antic-Scl-70, anticentromere)• Anti-cardiolipin antibodies and lupus anticoagulant• Serology for HIV, HCV, HBV, CMV and H1N1• Complete clotting test, with fibrinogen, fibrinogen degradation products and dimer D• Investigations for typical HUS-causing bacterial infections and Shiga toxin test (if clinically suspected) |
| Specific diagnostic tests | |
|---|---|
| • STEC infection | • Faecal sample in case of diarrhoea or rectal culture: STEC cultures (MacConkey for E. coli O157:H7); PCR for Stx genes O157:H7 and other serotypes, and other virus characteristics; ELISA and/or Vero cell tissue culture assay for Stx serum: anti-LPS antibodies for prevalent serotypes |
| • Pneumococcal infection | • Bacterial culture (generally) of sterile body fluids; DAT (Coombs test), viral test (respiratory), chest x-ray (pleural effusion as a characteristic in most cases), cytochemistry, and CSF culture in cases to pneumococcal meningitis |
| • Altered regulation of the complement | • C3, C4 (plasma/serum), AH50• FH, FI, FB (plasma/serum)• Anti-FH autoantibodies• Expression of superficial MCP in leukocytes (poly- or mononuclear leukocytes using a FACS test)• Mutation analysis in FH, FI, MCP, C3, FB±THBD |
| • ADAMTS13 deficiency (acquired or hereditary) | • Plasma activity of ADAMTS13 or dose (ELISA)±inhibitor |
| • Cobalamin metabolism: methylmalonic aciduria | • Amino acid chromatography in plasma/urine (hyperhomocysteinemia, hypomethioninemia; homocystinuria); organic acid chromatography in urine (methylmalonic aciduria)• Mutation analysis for the gene MMACHC |
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; ANA: antinuclear antibody; ANCA: Autoantibodies to neutrophil cytoplasmic antigens; CMV: cytomegalovirus; CSF: cerebrospinal fluid; DAT: direct antiglobulin test; DNA: deoxyribonucleic acid; ELISA: enzyme-linked immunoabsorption assay; FACS: fluorescence activated cell sorting; FB: complement factor B; FH: complement factor H; FI: complement factor I; HIV: human immunodeficiency virus; HUS: haemolytic uraemic syndrome; MCP: membrane cofactor protein; STEC: Shiga toxin-producing Escherichia coli; THBD: thrombomodulin; VHB: hepatitis B virus; VHC: hepatitis C virus.
Specific diagnostic tests: adapted from Loirat et al.2
In patients with TMA, tests results include thrombocytopenia (platelet count <150,000/mm3 or decrease >25% from baseline)20 and microangiopathic haemolytic anaemia (haemoglobin <10mg/dl with a negative direct Coombs test [though some patients with pneumococcal or H1N1-related HUS may show positive direct Coombs test],25 elevated LDH, decreased haptoglobin, reticulocytosis, and schistocytes).20,62 In a retrospective series of 50 patients with histological TMA, 44% had a normal platelet count.74 Consequently, diagnosis of TMA should be considered in patients with renal failure and elevated LDH, but without thrombocytopenia. For schistocytes, even though they can be found in most patients with renal disease, preeclampsia or mechanic valves, TMA can be diagnosed with a schistocyte count >1% provided that there are no other known causes.75 In contrast, the absence of schistocytes does not rule out a diagnosis of TMA.
High levels of serum creatinine, low glomerular filtration (GF) or the presence of proteinuria and/or haematuria11,20,69 are indicative of renal failure. A renal biopsy may be required for adult patients following acute renal failure to determine the aetiology, rule out other processes, and assess prognosis, although the indication and time for biopsies must be examined individually in patients with suspected TMA due to the risk of bleeding. In this sense, diagnostic renal biopsies are not recommended in patients conclusively diagnosed with aHUS (positive family history, recurrence, etc.). Among paediatric patients, the diagnosis is essentially made on the basis of clinical presentation, though renal biopsy may occasionally be required (especially in cases of secondary TMA or RT). Patients with clinical suspicion of TMA should always be examined by a nephrologist in light of the urgent treatment strategy required to ensure minimum irreversible renal damage.
Disseminated intravascular coagulation (DIC) is a syndrome that may be associated with several major laboratory and clinical findings related to TMA. DIC is characterised by a systemic activation of coagulation, secondary to several clinical conditions (sepsis, trauma or certain tumours), leading to thrombosis and bleeding, commonly involving renal function.76 Key test criteria based on coagulation tests for differential diagnosis of DIC and TMA are listed in Table 4.
Differential diagnosis between disseminated intravascular coagulation and thrombotic microangiopathy.
| DIC | TMA | |
|---|---|---|
| Platelet count | ↓ | ↓ |
| Fibrinogen | ↓ | Normal |
| Fibrinogen degradation products | ↑ | Normal |
| Dimer D | ↑ | Normal |
| Antithrombin | ↓ | Normal |
| Schistocytes | Present | Present |
| Haptoblogin | Normal | ↓ |
| Coagulation times | Long | Normal |
| Blood pressure | ↓ | ↑ |
DIC: disseminated intravascular coagulation; TMA: thrombotic microangiopathy.
A complete and detailed clinical history should be made for TMA patients, including personal and family history, triggering factors (drugs, infections), and a thorough physical examination. As opposed to previous considerations made several years ago, signs and symptoms of the different types of TMA are currently thought to be nonspecific and avoid the companion diagnostics between these two entities.1 The differentiation between HUS and TTP was classically based on clinical criteria, with HUS and TTP being diagnosed when renal involvement and neurological involvement were predominant, respectively. However, 50% of patients with TTP develop renal failure and 50% of patients with aHUS develop neurological abnormalities.37,77
Clinical features do not allow for a differentiation between STEC-HUS and aHUS as well, given that up to 30% of aHUS cases are developed following gastroenteritis12 or patients develop diarrhoea42 (a typical symptom of STEC-HUS). On the other hand, platelet count and the severity of renal involvement can actually guide companion diagnostics. Overall, TTP presents with severe thrombocytopenia (<20,000/mm3 in 73% of patients with acquired TTP)78 and moderate renal involvement, whereas aHUS usually presents with moderate thrombocytopenia (50–100,000/mm3) and severe renal involvement. This rule can be deemed a guide, but determination of ADAMTS13 activity and the Shiga toxin test are essential for an accurate differential diagnosis between TTP, STEC-HUS, and aHUS (Fig. 4). The diagnosis of STEC-HUS can be confirmed by the presence of the Shiga toxin or a positive culture of STEC in patients with TMA,28 while the activity of ADAMTS13 in plasma should be <5–10% in order to confirm the diagnosis of TTP.79,80 The diagnosis of the remaining cases should be directed towards aHUS,79 and so additional tests are required to rule out secondary TMAs. Test samples should be collected prior to PT.

Algorithm for the differential diagnosis of primary thrombotic microangiopathy. ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; aHUS: atypical haemolytic uraemic syndrome; HUS: haemolytic uraemic syndrome; LDH: lactate dehydrogenase; STEC: Shiga toxin producing Escherichia coli; TTP: thrombotic thrombocytopenic purpura. * Negative direct Coombs test. ** The Shiga toxin test/STEC is indicated when the patient has a history of digestive involvement or gastrointestinal symptoms. *** STEC infection can rarely trigger the underlying disease activity in some patients with aHUS.
Treatment for aHUS should involve two different strategies: on one side, supportive treatment measures aimed at managing the consequences of aHUS (acute renal failure, high blood pressure, anaemia, thrombocytopenia, etc.), and a targeted therapy to halt and revert TMA. Specific options for the management of aHUS will be reviewed in this section.
Plasma therapyPT can be delivered as plasma infusion (PI) and plasma exchange (PE). In PI, patients are given virus-inactivated, non-native fresh frozen plasma (FFP), adding functional complement regulators.81 In PE, a patient's plasma is replaced with FFP, which not only results in the administration of high doses of complement-regulating proteins, but also in the elimination of dysfunctional endogenous soluble complement inhibitors, minimising the risk of volume overload. In addition, anti-FH antibodies are also cleared in PE, together with potential inflammatory/thrombogenic factors involved in endothelial damage and platelet hyperaggregation. The treatment of choice recommended for episodes of aHUS traditionally consisted of early and intensive PE at high volumes and of variable frequency based on disease activity. PIs are usually ineffective except in a few patients with complete deficiency of FH82 (circulating levels of complement proteins are normal in most patients). Overall, PT is not considered effective in patients with isolated MCP mutations, as this is a non-circulating protein attached to the cell membrane, with virtually all patients relapsing following an episode of aHUS irrespective of the use of PT.12
Even though no prospective clinical trials are available, PT has empirically been the treatment of choice in aHUS for years as mortality in patients with TTP-HUS decreased over the last 3 decades. Table 5 summarises the results of the largest international registry of PT in patients with aHUS (International Registry of Recurrent and Familial HUS/TTP), including 273 patients diagnosed between 1996 and 2007.12 Complete haematological and renal recovery rates with PT in this registry are generally below 50% (except for patients with mutations in THBD and MCP), and particularly low rates in patients with mutations in FH and FI (5 y 12.5%).12 Mortality and/or outcome of ESRF are generally high in 3 out of 4 patients with FI mutations. Some papers prove that early intensive PE is essential to prevent patients from developing aHUS, and maintenance can prevent disease recurrence and ESRF,11,81 though the most effective management strategy is still not known, nor is the long-term impact on renal function.
Prognosis of patients with atypical haemolytic uraemic syndrome treated with plasma infusion or plasma exchange.
| Remission | Death or end-stage renal failure | |
|---|---|---|
| CFH | 63% (complete: 5%; partial: 58%) | 37% |
| CFI | 25% (complete: 12,5%; partial: 12,5%) | 75% |
| C3 | 57% (complete: 43%; partial: 14%) | 43% |
| THBD | 88% (complete: 62%; partial: 25%) | 13% |
| Anti-FH antibodies | 75% (complete: 25%; partial: 50%) | NA |
| MCP | 97% of treated patients (complete: 90%; partial: 7%) y 100% of non-treated patients | NA |
Anti-FH: anti-complement factor H antibodies; CFH: complement factor H gene; CFI: complement factor I gene; MCP: membrane cofactor protein gene; NA: not available; complete remission: haematological and renal function normalisation; partial remission: haematological normalisation and renal sequels; THBD: thrombomodulin gene.
Adapted from Noris et al.12
Concomitant immunosuppression and PT may improve outcomes in patients with anti-FH antibodies.19,83,84 High antibody titres are correlated with a higher risk of relapse and renal sequels in these cases.19 Even though further trials are required to conclude on how anti-FH antibodies are developed by patients with aHUS, the fact that the vast majority of these patients have complete deficiency of FHR1 leads to the idea that these antibodies are really targeted against the FHR1 protein, and that the anti-FH activity is a cross reaction resulting from the remarkable homology existing between these 2 proteins. This possibility is a warning of the potential anti-FHR1/anti-FH sensitisation in homozygotes for CFHR3-CFHR1 deletion by the exposure to exogenous FHR1, discouraging the use of PI in these individuals.
The potential complications of PI are anaphylactic reactions to FFP, hypervolemia, high blood pressure, heart failure, or hyperproteinaemia. The main complications of PE are obstructed venous access (6%), low blood pressure (5%), and allergy (4%)85, with a higher frequency among paediatric patients.85 A study conducted in 71 paediatric patients with aHUS (59 treated with PE) showed that 80% of children had some renal sequels within one month of follow-up, 17% were dialysis-dependant, and 31% developed catheter-related complications.86
EculizumabEculizumab is a humanised monoclonal IgG2/4 kappa antibody that binds to the C5 complement protein with high affinity, blocking the excision into C5a and C5b, and preventing the formation of the C5b-9 complex of the terminal complement (membrane attack complex) (Fig. 3).4 In aHUS, the dysregulation of the alternative complement pathway leads to the uncontrolled activation of C5, causing a damage to self structures via the formation of the membrane attack complex. This process is rapidly and sustainably reduced as a result of the blockade of the terminal complement pathway by Eculizumab. A large number of patients with aHUS have shown a good clinical response to the drug (Table 6).
Published cases of patients with atypical haemolytic uraemic syndrome receiving Eculizumab (last updated in April 2014).
| Patients with aHUS in native kidneys | |||||
|---|---|---|---|---|---|
| Reference | Mutation | Response to plasma therapy | Baseline serum creatinine, μmol/l | Patient outcome | Last serum creatinine value, μmol/l |
| 121,122 | Unidentified | Resistant to plasma exchange | 265 | Remission after 3 years | 35 |
| 123 | CFH | Partially sensitive to plasma exchange | 80 | Remission after 10 weeks | 26 |
| 124a | Unidentified | Resistant to plasma exchange | 690 | Recurrence after 2 weeks | End-stage renal failure |
| 125a | Unidentified | Resistant to plasma exchange | ∼310 | Recurrence after 2 weeks | End-stage renal failure |
| 126 | CFH S1191L V1197A | Resistant to plasma infusion | 108 | Remission after 15 months | 44 |
| 127 | CFIp.A258 T | Resistant to plasma exchange | 610 | Remission after 7 months | 230 |
| 38,128b | Unidentified | No plasma therapy | 600 | Remission after 6 months | 125 |
| 129 | CFHC611Y | Intolerance to plasma exchange | ∼230 | Remission after 24 months | ∼100 |
| 130 | Unidentified | Resistant to plasma exchange | ∼325 (dialysis) | Remission after 9 months | ∼80 |
| 131 | CFH | Resistant to plasma exchange | ∼310 (dialysis) | Remission after 18 months | ∼75 |
| 132 | MCPc.286+ 1G>C | Resistant to plasma exchange | Dialysis | Haematological normalisation | End-stage renal failure |
| 133 | Unidentified | Resistant to plasma infusion | Continuous hemodiafiltration | Remission after one year | 18 |
| 134 | CFH3355 G>a; Asp1119Asn; SCR19 | Resistant to plasma exchange | Dialysis | Remission>2.5 years | 26 |
| 43a | Unidentified | Sensitive to plasma exchange | Dialysis | Resolution of thrombocytopenia and skin lesions | Dialysis |
| 135 | CFH | Partially sensitive to plasma exchange | 723 (dialysis) | Remission | Dialysis-free |
| 136c | Unidentified | − | ∼247 (dialysis) | Remission after 6 months | Dialysis-free |
| 137 | C3 | Resistant to plasma therapy | Dialysis | Remission after 2 years | Dialysis |
| 138 | CFH3514G>T | Resistant to plasma therapy | ∼222 (dialysis) | Remission after one year | 117 |
| 139 | C3c3466G>A | Resistant to plasma exchange | Dialysis | Remission>11 months | 115 |
| 140a | C3 | – | Dialysis | Remission | Dialysis-free |
| 141c | CFBc.967A>C; p.Lys323Gln | – | 20 | Favourable outcome at 6 months, in spite of the persistence of slightly increased plasma levels of LDH y C5b-9 | Normal levels |
| 142 | CFHp.Arg53Cys; c.157C.T | Resistant to plasma exchange | Normal levels | Remission | Normal levels |
| 143c | CFHp.Lys1186ThrCFIp.Ile322Thr | Partially sensitive to plasma exchange | Dialysis | Remission | 75 |
| 144 | CFIc.786delA | Resistant to plasma exchange | Dialysis | Remission | 88 |
| Renal transplant patients | ||||||
|---|---|---|---|---|---|---|
| Preventive use of Eculizumab | ||||||
| Reference | Mutation | Previous transplants (number) | Response to plasma therapy | Baseline serum creatinine, μmol/l | Patient outcome | Last serum creatinine level, μmol/l |
| 107 | CFH W1183C | No | Sensitive to plasma exchange | ∼45 | No recurrence | 44 |
| 108 | CFH E1198stop | No | No plasma therapy | Dialysis | No recurrence | Normal |
| 109 | CFH/CFHR1hybrid gene | No | Sensitive to plasma exchange | Dialysis | No recurrence | 80 |
| 110 | CFH/CFHR1 hybrid gene | No | Sensitive to plasma exchange | Dialysis | No recurrence | Normal |
| 111 | CFH/CFHR1 hybrid gene | No | No plasma therapy | Dialysis | No recurrence | 79 |
| 112 | CFH c.3497C9T | No | Resistant to plasma exchange | Dialysis | No recurrence | 76 |
| Use of Eculizumab for the treatment of post-transplant aHUS recurrence | ||||||
|---|---|---|---|---|---|---|
| 145a | CFH Y475S | Yes (1) | Resistant to plasma exchange | 132 | Graft loss | NS |
| 146,147 | C3 R570Q | Yes (1) | Sensitive to plasma exchange | 320 | 2 recurrences in casesof delayedEculizumab | 230 |
| 97 | Unspecified | No | Resistant to plasma exchange | 323 | Remission | 238 |
| 98 | CFH S1191L | Yes (2) | Intolerance to plasma exchange | 131 | Remission | 130 |
| 148a | Unidentified | No | Resistant to plasma exchange | 415 | Graft loss | NS |
| 42 | CFH | Yes (1) | Resistant to plasma exchange | 500 | Remission | 62 |
| 69 | C3 R570W | Yes (2) | Partially sensitive to plasma exchange | 220 | Remission | 115 |
| 99 | CFH E3514Stop | No | Partially sensitive to plasma exchange | 565 (dialysis) | Remission | 229 |
| 149 | Unidentified | Yes (1) | Resistant to plasma exchange | 449 (dialysis) | Recurrence 5 months following withdrawal of Eculizumab. Graft loss | NS |
| 43 | CFH | No | Partially sensitive to plasma infusiond | 220 | Remission (disappearance of skin lesions) | 209 |
aHUS: atypical haemolytic uraemic syndrome; CFB: complement factor B gene; CFH/CFHR1: complement hybrid gene resulting from CFH/CFHR1 conversion; CFH: complement factor H gene; CFHR1: complement factor H-related protein 1 gene; CFI: complement factor I gene; HUS: haemolytic uraemic syndrome; LDH: lactate dehydrogenase; TMA: thrombotic microangiopathy; NE: not specified.
The efficacy and safety of Eculizumab in aHUS were initially assessed in two phase II, prospective, multicentre trials, including 37 patients older than 12 years of age and with primary or recurrent disease following RT, who received Eculizumab for 26 weeks, followed by long-term extension periods.5 Seventeen patients with aHUS (mean time from diagnosis: 9.7 months) with evidence of progressive TMA following ≥4 sessions of PT the week before their inclusion (C08-002) were enrolled in the first study. The second study recruited 20 patients (mean time from diagnosis: 48.3 months) receiving PT (1 session every 2 weeks and 3 sessions a week) where no decrease >25% was reported in platelet count for at least 8 weeks prior to first dosing of Eculizumab (C08-003). Genetic or anti-FH antibody mutations were observed in 76% and 70% of patients from the first and the second study, respectively. Primary outcomes in both studies were: (a) inhibition of complement-mediated TMA (study 1: increased platelet count; study 2: TMA-free patient ≥12 weeks [no decrease in platelet count >25%, no PT and no dialysis]), and (b) haematological normalisation (≥2 normal consecutive measurements of platelets and LDH, with a minimum interval of 4 weeks). Secondary outcomes included change in the rate of daily interventions for TMA (PI or PE sessions, dialysis, or both, per patient/day), renal function, quality of life, safety and tolerability. Primary outcomes reported at 26 weeks and in extension studies are described in Table 7. With regard to primary outcome, following 26 weeks of treatment, in study 1 treatment with Eculizumab was associated with a significant increase in the number of platelets from baseline (p<0.001) and a rate of haematological normalisation of 76%. In study 2, 80% of patients were free of TMA episodes following 26 weeks of treatment with Eculizumab and 90% had haematological normalisation. In terms of secondary outcomes, treatment with Eculizumab at 26 weeks was associated with a significant reduction in the rate of daily interventions for TMA vs. baseline (p<0.001), as well with a continuous improvement of estimated GFR (+32ml/min/1.73m2 [p=0.001 vs. baseline] and +6ml/min/1.73m2 [p<0.001 vs. baseline] in studies 1 and 2, respectively), a decrease in proteinuria (p<0.05) and a reduced need for dialysis. Also, the earlier Eculizumab is introduced in trials (less time of evolution between the clinical manifestation of aHUS and the drug), the more significant the improvement of the estimated GFR (p<0.05). The results of extension trials at 1 and 3 years prove that long-term treatment with Eculizumab is associated with maintenance or progressive improvement of the haematological response and renal function.87,88 All these positive results with Eculizumab were observed irrespectively in patients with or without genetic or anti-FH antibody abnormalities.
Main results from prospective studies with Eculizumab in patients with aHUS.
| C08-002 (n=17) | C08-003 (n=20) | C10-003 (n=22) | C10-004 (N=41) | |||||
|---|---|---|---|---|---|---|---|---|
| Week 26 | Week 64 | Week 100 | Week 26 | Week 62 | Week 156 | Week 26 | Week 26 | |
| Change in platelet count from baseline (×109/l), meanp value vs. baseline | +73a<0.001 | +910.001 | +97<0.0001 | +5NS | NA | NA | +164<0.0001 | +135<0.0001 |
| Platelet count normalisation,b number of patients (%) | 14 (82) | 15 (88) | NA | NA | NA | NA | 21 (95) | 40 (98) |
| No TMA events,c number of patients (%) | 15 (88) | 15 (88) | 15 (88) | 16 (80)a | 17 (85) | 19 (95) | 21 (95) | 37 (90) |
| Haematological normalisation (complete haematological response)d, number of patients (%) | 13 (76) | 15 (88) | 15 (88) | 18 (90) | 18 (90) | 18 (90) | 18 (82) | 36 (88) |
| Daily intervention rate for TMAe (number of events/patients/day)Before Eculizumab, meanOn Eculizumab, meanp value vs. “before Eculizumab” | 0.880<0.001 | 0.880<0.001 | NA | 0.230<0.001 | 0.230<0.001 | NA | NA | NA |
| Complete response for TMA,f number of patients (%) | 11 (65) | 13 (76) | NA | 5 (25) | 7 (35) | NA | 14 (64)a | 30 (73)a |
| Outcome of estimated GFR (ml/min/1.73m2)p value vs. baseline | +320.001 | +32<0.001 | +38≤0.05 | +6<0.001 | +90.003 | +4NS | +64<0.0001 | +29<0.0001 |
| Reduction≥25% in serum creatinine, number of patients (%) | 11 (65) | 13 (76) | 13 (76) | 3 (15) | 7 (35) | 11 (55) | 16 (73) | NA |
| Increased estimated GFR≥15ml/min/1.73m2, number of patients (%) | 8 (47) | 9 (53) | 10 (59) | 1 (5) | 3 (15) | 8 (40) | 19 (86) | 22 (54) |
| No dialysis, number of patients/number of patients in dialysis at the start of treatment (%) | 4/5 (80) | 4/5 (80) | NA | 0/2 (0) | 0/2 (0) | NA | 9/11 (82) | 20/24 (83) |
| Improvement in CRD in at least one stage, number of patients (%) | 10 (59) | 11 (65) | 13 (76) | 7 (35) | 9 (45) | 12 (60) | 17 (77) | 26 (63) |
| Reduction of proteinuria in at least one grade in patients with baseline proteinuria grade≥1, number of patients/total number of patients | 12/15 | 9/11 | NA | 6/11 | 7/9 | NA | NA | NA |
| Improvement in quality of life (change in questionnaire scoring), meangp value vs. baseline | +0.32<0.001 | +0.30<0.001 | +0.29<0.0001 | +0.10<0.001 | +0.13<0.001 | +0.16≤0.001 | +19.7<0.0001 | +0.230.003 |
aHUS: haemolytic uraemic syndrome; CRD: chronic renal disease; EQ-5D: EuroQoL Group 5-Dimension Self-Report Questionnaire; FACIT-F: Functional assessment of chronic illness therapy-fatigue; GFR: glomerular filtration rate; LDH: lactate dehydrogenase; NA: not available; NS: not significant; PE: plasma exchange; PI: plasma infusion; TMA: thrombotic microangiopathy.
Overall, tolerability of Eculizumab was good, and severe treatment-related adverse effects were only reported in 4 patients in each study, probably in the context of the background condition, aHUS (study 1: malignant hypertension,2 severe hypertension, and asymptomatic bacteriuria; study 2: influenza infection, peritonitis, venous sclerosis at the time of the infusion, and fever Q). Increased risk of infection by encapsulated germs, especially Neisseria meningitides, results from the mode of action of Eculizumab, and so all patients were vaccinated against Neisseria (tetravalent vaccine) 14 days before treatment and/or received antibiotics, with no reports of meningitis. Only one patient died of digestive bleeding 3 years later (unrelated to Eculizumab).
The safety and efficacy of Eculizumab during pregnancy has recently been studied in patients with paroxysmal nocturnal haemoglobinuria. Data from 61 women with 75 pregnancies while on treatment with Eculizumab suggest a good tolerability profile of Eculizumab during pregnancy, with high rates of foetal and maternal survival (96% and 100%, respectively). Eculizumab was observed in 7 out of 20 blood samples of umbilical cord and in none of the 10 samples of human milk.89
Two phase III, multicentre, prospective, open-label trials of Eculizumab in aHUS are currently ongoing, one in adult patients (n=41; C10-004)90 and the other in paediatric patients (n=22; C10-003 22).91 Unlike previous trials, these new studies primarily involve patients who have been recently diagnosed with aHUS (73%). In the adult trial, mean time from diagnosis to inclusion was ∼3 weeks, whereas mean time from clinical onset to Eculizumab was 2 weeks (15% of patients had not been treated with PT prior to Eculizumab).90 The paediatric trial involves patients with aHUS ≤18 years with a mean time from diagnosis to inclusion of ∼2 weeks and a mean time from clinical onset to Eculizumab of ∼1 week (55% had not been treated with PT).91 Results from these 26-week trials confirm the significant haematological and renal function improvements seen in previous studies, as well as the benefits of early use of Eculizumab (Table 7).90,91 The safety profile was similar, although two patients developed meningococcal meningitis (5%) in the adult study. Both infections were well-managed, and one of the patients continued treatment with Eculizumab. Survival in patients from both studies was 100%.
Blood samples from adult patients enrolled in the C10-004 trial were collected at baseline (before treatment with Eculizumab) and in subsequent visits up to Weeks 49–54 in order to measure the impact of Eculizumab on biomarkers related to TMA and endothelial damage in aHUS.92 Patients with aHUS (irrespective of the presence or absence of related mutations, PT, or haematological values) showed activated complement system and inflammation, coagulation, activation, endothelial damage, and renal injuries prior to treatment. Eculizumab led to normalisation of complement activation-related biomarkers and significantly reduced biomarkers of inflammation, thrombotic risk, endothelial and organic damage. Eculizumab also reduced biomarkers related to the activation of the alternative complement pathway and endothelial activation, although they did not completely return to normal. These findings underline the persistent chronic of complement activation in patients with aHUS and a continuous risk of TMA and potential organ damage.92
There is also a retrospective study including 19 paediatric patients with aHUS receiving Eculizumab in a clinical setting for a mean time of 28 weeks (C09-001).3 Eighty nine percent of patients in this study had a normalised platelet count and 68% remained free of TMA episodes, although most of them were under haematological remission with prior treatment with PE. The rate of interventions for TMA was reduced from 0.3 per patient/week to 0 (p<0.0001). GFR increased to ≥15ml/min/1.73m2 in 47% of patients and 50% required no dialysis at all. Pyrexia (47%), diarrhoea (32%), and upper respiratory tract infections (32%) were the most common adverse effects.
Recommendations for the management of the atypical haemolytic uraemic syndromeEver since Eculizumab was approved for the treatment of aHUS by the EMA and the Spanish Agency for Drugs and Health Products (AEMPS) in 2011,3 management and prognosis among patients with aHUS have substantially improved, as the drug was approved for use as first-line therapy. Recommendations for the treatment of aHUS made by the authors of this document based on available evidence and cumulative clinical experience are summarised below.
Treatment of atypical haemolytic uraemic syndromeIn view of technical difficulties of PT in paediatric patients (by body size) and potential complications, in addition to the superiority of Eculizumab for the recovery of renal function (and the resulting improved prognosis), early first-line treatment with Eculizumab is highly recommended in this population, therefore avoiding the use of PE. Consequently, early administration of Eculizumab is recommended as treatment of choice in first line in paediatric patients with suspected aHUS (Fig. 5).91 Eculizumab should be initiated earlier in adult patients with suspected aHUS following PE.90 PE is only recommended in adults when diagnosis is unclear. The use of Eculizumab may only be disregarded in patients with haematological complete recovery and improved renal function following PE. In this study, the aHUS French Study Group recommended switching the patient to Eculizumab if platelet count or LDH levels do not return to normal following the fifth PE, or reducing plasma creatinine ≥25%.93 Eculizumab is the treatment of choice recommended whenever the diagnosis of aHUS is unequivocal (positive personal or family history or disease recurrence following RT). Early administration ensures reversibility of haematological parameters and prevents renal injuries.
, Eculizumab should be use earlier as treatment of choice.")
Treatment for atypical haemolytic uraemic syndrome. PE: plasma exchange; aHUS: atypical haemolytic uraemic syndrome. * When the diagnosis of aHUS is unequivocal (positive personal or family history or disease recurrence following renal transplant), Eculizumab should be use earlier as treatment of choice.
All patients should be vaccinated against N. meningitides (preferably with conjugate tetravalent vaccines against serotypes A, C, Y, and W135, and serotype B) prior to treatment with Eculizumab. If treatment with Eculizumab cannot be delayed until vaccine response, associated treatment with antibiotics against N. meningitides may be initiated and antibiotic prophylaxis may be established3 as per the hospital protocol. Given the higher frequency of invasive meningococcal infection among paediatric patients, and the absence of serotype B protection (currently the most prevalent following systemic vaccination in the population for other serotypes), this age group should continue to use antibiotic prophylaxis, including penicillin or amoxicillin, in combination with Eculizumab,2 although these prophylaxis protocols may be adjusted following the recent availability of the new serotype B vaccine. The maintenance of antibiotic prophylaxis in adult patients receiving Eculizumab is at the physician's own discretion and should be individually assessed. Continued antibiotic prophylaxis should be considered in immunodepressed patients receiving Eculizumab as a result of the lower response to the vaccine among these patients (particularly among those receiving renal transplants). In paediatric patients, Haemophilus influenzae and pneumococcal vaccines are also required, together with strong compliance of local effective recommendations on compulsory vaccines for each age group.
If response to Eculizumab is good, treatment should be maintained indefinitely as recommended in the summary of product characteristics.3 No recommendations can be made so far on the right treatment duration, though increasing experience with drug use may help better define this as well as treatment strategies in the future.
Withdrawal and/or individual dose titration of Eculizumab may be considered for specific cases, but only among low-risk patients (with isolated mutation in MCP and negative family history), always individually and following at least 12 months of treatment.94,95 Patients who withdraw treatment as per clinical indication should be carefully monitored for at least 12 weeks for potential abnormalities suggestive of TMA and/or relapse.3,96 In these patients immediate readministration with Eculizumab should be considered.3,96
Eculizumab should be maintained for at least 3 months in patients with aHUS and acute renal failure requiring dialysis in order to assess the improvement of renal function. Progressive increase of diuresis with good pressure management are positive parameters guiding the management of the TMA process and improvement of renal injuries. Renal biopsies in patients receiving dialysis may help decision-making associated with treatment continuation. If treatment fails in patients and they still require dialysis for renal failure, Eculizumab should be withdrawn, except in patients with systemic disease manifestations, where treatment continuation should be assessed individually.
Whenever PT is considered for a patient with aHUS, PE should preferably be performed with FFP replacement (1.5 per plasma volume [60–75ml/kg] per session to add complement factors). Sessions should be performed until platelet count returns to normal, end of haemolysis ends, and sustained improvement of renal function for several days occurs. Five weekly sessions should be conducted thereafter for the first 2 weeks and 3 weekly sessions for the next 2 weeks, while PE continuation should be individually assessed.2,82
Patients with aHUS who develop anti-FH antibodies while on PT should use concomitant immunosuppressants to prevent antibody formation.19,73,83,84 Treatment response among these patients should be monitored based on the outcome of antibody titres.73
General supportive measures are necessary to ensure acceptable conditions for patients until TMA is managed. High blood pressure is common among patients with aHUS and should be treated with angiotensin II blockers (ACEI or AIIRA). Volemic control is also vital due to common hypervolemia and acute risk of pulmonary oedema. Transfusions of red blood cells concentrates and/or the use of erythropoiesis-stimulating factors should be considered for the treatment of anaemia. Platelet transfusions should be reserved for cases of severe low platelet counts (<30,000/mm3) or exceptionally for severe bleeding and/or prior to invasive procedures with risk of bleeding, as the TMA phenomenon may become worse. Potential agents leading to aHUS should also be identified and managed. Paediatric patients with aHUS should be referred to specialised centres for paediatric nephrology including expert staff and an intensive care unit for paediatric patients in order to ensure adequate treatment.
Atypical haemolytic uraemic syndrome and transplantationTreatment for the recurrence of atypical haemolytic uraemic syndrome in renal transplantationPrimary clinical criteria for the differential diagnosis of the different types of TMA in RT and an algorithm for the management of these entities are described in Table 8 and Fig. 6, respectively. Diagnosis in patients receiving renal transplantation and developing TMA, with a history of aHUS episodes before transplantation, should be directed to disease recurrence (ruling out other potential causes).
Differential diagnosis of TMA in renal transplant patients.
| TMA de novo | Recurrence of aHUS | |
|---|---|---|
| History of HUS/TMA | No | Yes |
| Systemic involvement | No | Common |
| Intensity of clinical picture | Mild | Severe |
| Onset | Progressive | Sudden |
| Intensity of haematological TMA | Low | High |
| Causative/triggering agents | CNI, mTORiViral infectionsHumoral rejection | Triggering factors are not always found |
| Reversibility | Yes | No. Graft loss |
aHUS: atypical haemolytic uraemic syndrome; CNI: calcineurin inhibitors; HUS: haemolytic uraemic syndrome; mTORi: mammalian target of Rapamycin inhibitors; TMA: thrombotic microangiopathy.
Adapted from Zuber et al.68
; IS: immunosuppression; TMA: thrombotic microangiopathy; AHR: acute humoral rejection; PE: plasma exchange; HUS: haemolytic uraemic syndrome; aHUS: atypical haemolytic uraemic syndrome; STEC: Shiga toxin-producing Escherichia coli. a STEC-HUS recurrence following renal transplantation is very rare.")
Treatment for thrombotic microangiopathy in renal transplantation. CMV: cytomegalovirus; CNI: calcineurin inhibitors; mTORi: mTOR inhibitors (mammalian target of Rapamycin); IS: immunosuppression; TMA: thrombotic microangiopathy; AHR: acute humoral rejection; PE: plasma exchange; HUS: haemolytic uraemic syndrome; aHUS: atypical haemolytic uraemic syndrome; STEC: Shiga toxin-producing Escherichia coli. a STEC-HUS recurrence following renal transplantation is very rare.
Treatment for the recurrence of aHUS in patients receiving renal transplants should be performed under the same terms as in aHUS of native kidneys by means of the early use of Eculizumab.5,42,43,69,90,91,97,100 In view of the induced immunosuppression status of transplanted patients (chronic immunosuppression), vaccination against N. meningitides is also recommended to assess the maintenance of antibiotic prophylaxis while on treatment with Eculizumab.
In patients with progressive TMA in the first study of Eculizumab, renal function recovery following administration was significantly improved in the long term in nontransplanted patients (with aHUS in native kidneys) compared to patients receiving renal transplantation.5 This result may be related to the fact that transplanted patients enrolled in the study received Eculizumab later than nontransplanted patients (mean time from clinical onset to enrolment: 1.71 and 0.67 months for both types of patients, respectively). This observation stresses the need to early use Eculizumab for aHUS recurrence in RT.
Prophylaxis for the recurrence of the atypical haemolytic uraemic syndrome following renal transplantationPerspectives of RT in patients with ESRF secondary to aHUS receiving dialysis have significantly changed in recent years, especially in patients with high risk of disease recurrence following transplantation (patients with risk mutation and/or relapse of aHUS). The use of Eculizumab has made treatment options available for these patients for whom RT was contraindicated because of the high rate of relapse and the risk of renal graft loss. There are currently three treatment options to prevent aHUS recurrence following RT: (a) a liver–kidney combined transplantation; (b) simple RT together with prophylaxis with PT, and (c) simple RT with prophylactic Eculizumab.
Over 20 cases of patients with aHUS and mutations in genes coding for complement factors primarily synthesised in the liver (FH, FB or FI) and receiving liver transplants (isolated or combined with RT) have been recently reported so as to avoid the consequences of the genetic defect and prevent disease recurrence. This strategy, combined with the perisurgical use of Eculizumab or plasma (to eliminate dysfunctional complement factors during surgery and add enough functional factors until liver function is recovered), was successful in several occasions, with good liver function and no recurrence of aHUS during follow-up.70,101–105 Nonetheless, given the potential morbi-mortality related to liver transplants,2 and the challenge posed by the availability of organs and safer alternatives, liver–kidney transplants should only be considered in selected patients with aHUS as a second option. Isolated liver transplants are also not recommended for patients with aHUS and functional kidneys, as chronic immunosuppression risks outweigh risks related to long-term use of Eculizumab.68
Prophylaxis with PT in patients with ESRF secondary to aHUS and receiving simple RT has been associated with positive outcomes in terms of disease recurrence prevention in several papers.68 Yet, recurrence of aHUS has been reported following RT in patients at risk, including the use of intensive PT.68,100 On the other hand, factors including the potential increased risk of disease recurrence with progressive spacing of PT sessions106 or the impact on QoL in patients resulting from long-term PT (mostly related to sustained vascular access) are restrictive of this strategy.68
Several positive experiences with the use of prophylaxis with Eculizumab among paediatric patients receiving FH prior to RT from a cadaveric donor have been reported in the last years (Table 6),107–111 therefore suggesting that RT associated with prophylactic Eculizumab is an effective and well-tolerated option for these patients.68 Zuber et al.100 have recently published a series including 9 patients receiving prophylaxis with Eculizumab for aHUS recurrence following RT.100 This series involves 6 paediatric patients and 3 adults with various mutations in the alternative complement pathway (5 in FH, one in C3 and 3 patients with hybrid genes resulting from the non-homologous recombination between CFH and CFHR1). Two patients receiving post-RT PE were later switched to Eculizumab, two patients received Eculizumab from the week before transplantation (unrelated living donor; urgent transplant from cadaveric donor) and the remaining 5 received Eculizumab immediately following transplantation. The other 8 patients had a positive outcome without recurrence following a mean follow-up of 14.5 months (mean creatinine: 71.6±44.8μmol/l), except for one case of early thrombosis leading to graft loss. Blasco Pelicano et al.112 reported the first use of prophylactic Eculizumab in our country in an adult female patient with FH mutation and a favourable outcome, without signs of relapse following 3 years of RT.112 Patients with ESRF secondary to aHUS who are eligible for RT should therefore use prophylactic Eculizumab as a first option for the prevention of aHUS recurrence following transplantation.68,93Fig. 7 includes an algorithm for the previous assessment and management of aHUS patients who are eligible for RT.
, together with screening for anti-FH antibodies. b RT with an unrelated living donor will only be considered if Eculizumab is available.")
Recommendations for the management of patients with ESRF secondary to aHUS who are eligible for RT. Adapted from Zuber et al.68 aHUS: atypical haemolytic uraemic syndrome; CFB: complement factor B gene; CFH/CFHR1: complement hybrid gene resulting from CFH/CFHR1 conversion; CFH: complement factor H gene; CFHR1: complement factor H related protein 1; CFI: complement factor I gene; DGKE: Diacylglycerol Kinase, Epsilon 64kDa; ESRF: end-stage chronic renal failure; FH: complement factor H; MCP: membrane cofactor protein gene; RT: renal transplantation. CKTD: chronic terminal kidney disease; KT: kidney transplant. a Determination of plasma levels of C3, C4, FH, FI and FB, as well as MCP expression in peripheral leukocytes; complete genetic study to detect known complement mutations (and risk polymorphisms), together with screening for anti-FH antibodies. b RT with an unrelated living donor will only be considered if Eculizumab is available.
There are also positive experiences on liver–kidney transplants using prophylactic Eculizumab,105 although as explained before, liver–kidney transplants should be considered as a second option for selected patients with aHUS.
Living donor transplants have traditionally been contraindicated in patients with aHUS in view of the high rates of disease recurrence and graft loss, and the risk of undetectable mutations in the complement system of the donor potentially resulting in the subsequent development of aHUS.1,66,113 Progress made so far in the field of genetic diagnosis and the availability of Eculizumab allow for consideration of related living donor transplantation as a valid option for patients with aHUS. A complete genetic-molecular exam should always be performed in donors, who will only be eligible based on mutations identified in the patient and absent in the donor. However, if the related donor and the recipient share any genetic factor of susceptibility to aHUS, or if no mutations are observed both in the recipient or the donor, no genetically matched living donor transplantation should be performed.93 Also, a living donor transplantation should only be considered if Eculizumab is available.
No specific protocols based on prospective trial on immunosuppression to reduce the risk of post-RT relapse are available. In general, calcineurin inhibitors114 and mTOR inhibitors68,115 are thought to be related to post-RT TMA, with a synergic effect in the combination of both drugs.116 These drugs should therefore be carefully used in patients receiving renal transplants due to ESRF secondary to aHUS. Guidelines on immunosuppression based on belatacept could be followed depending on the immunological risk of each patient, although no conclusive data are available in the literature as to the best immunosuppressive strategy for the risk population.
Conclusions- •
aHUS results from genetic or acquired dysregulation of the activated alternative complement pathway on cell surfaces leading to systemic TMA. Several mutations and polymorphisms have been described in recent years in genes from certain complement factors associated with this dysregulation.
- •
Clinical presentation comprises the triad of nonimmune microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal dysfunction, associated with common extra-renal manifestations. Before Eculizumab became available, aHUS was generally associated with increased mortality and/or progression to ESRF, as well as to increased recurrence following RT.
- •
Diagnosis should point to aHUS if TMA is clinically suspected and provided that the Shiga toxin/STEC test is negative, plasma ADAMTS13 is >5–10% and if secondary forms of HUS have been ruled out.
- •
Eculizumab is a monoclonal antibody inhibiting C5 activation and the formation of the membrane attack complex, responsible for damage to self structures in aHUS. In prospective studies including patients with aHUS, Eculizumab effectively prevented the TMA process and was associated with long-term significant haematological and renal function improvements.
- •
The authors of this paper recommend the early use of Eculizumab for aHUS in paediatric and adult patients with clinically suspected aHUS in native kidneys, and aHUS recurrence following RT or post-RT de novo aHUS.
- •
Prophylaxis with Eculizumab is recommended for the prevention of aHUS recurrence in patients with secondary ESRF receiving RT from a living or cadaveric donor.
- •
Eculizumab should be considered in patients with secondary TMA refractory to usual treatment.
This document has been approved by the Spanish Transplant Society (SET), the Spanish Society of Nephrology (SEN), and the Spanish Association of Paediatric Nephrology (AENP).
Sources of funding and acknowledgementsResearch carried out by Dr. Rodríguez de Córdoba has been funded by the Ministry of Economy and Competitiveness (SAF2011-26583), the Community of Madrid (S2010/BMD2316), the European Union (EURenOmics), the Fundación Renal Iñigo Álvarez de Toledo, and the CIBER of rare diseases.
The authors would like to thank Alexion Pharmaceuticals for the logistical support provided for meetings held by the Spanish Group for atypical haemolytic uraemic syndrome, as well as for the editorial assistance provided by Ogilvy Healthworld.
Conflicts of interestDr. Ariceta, Dr. Blasco, Dr. Campistol, Dr. Praga, Dr. Rodríguez de Córdoba, Dr. Valdés, Dr. Vilalta and Dr. Román have provided consultancy and teaching assistance to Alexion Pharmaceuticals. Dr. Torra has been a member of the expert committees in aHUS sponsored by Alexion Pharmaceuticals. Dr. Espinosa has taken part in clinical trials sponsored by Alexion Pharmaceuticals. None of these activities have influenced the development and interpretation of this manuscript, the authors being solely responsible for the contents herein displayed.
The management of secondary forms of HUS should be based on the treatment of the primary aetiology of the disease and the administration of PE. As explained, there is an increasing number of reports in the literature from patients with secondary HUS showing good response to Eculizumab, therefore suggesting that the complement plays an important role in the development of TMA in certain patients with secondary HUS.30–34,117–120 Consequently, the potentially temporal administration of Eculizumab should be considered as salvage therapy in selected patients with secondary HUS refractory to initial treatment measures. Also, screening for complement gene mutations in patients with secondary HUS points out that the underlying disease is actually aHUS, and so these patients should be treated as per the guidelines described in “Recommendations for the management of the atypical haemolytic uraemic syndrome”.
Although unnecessary to the clinical diagnosis of aHUS, a research of the complement including plasma levels of all factors should be conducted, together with a complete genetic analysis of all affected patients. Samples should be collected prior to treatment initiation, including plasma exchange, and should be sent to a reference laboratory (Table 9). As mutations have been identified in the complement system of patients with secondary STEC-HUS and HUS, genetic tests should also be individually conducted in these patients to study the potential association with complement/aHUS abnormalities.2
Protocol for sample collection for complement tests in patients with atypical haemolytic uraemic syndrome.
| • If samples are to be sent to a reference laboratory, we strongly recommend to contact the laboratory before and follow the instructions on samples required for assays |
| • If no reference laboratory is available and samples are to be stored for further tests, samples to be collected are: |
| • 10ml of blood with EDTA |
| • 10ml of coagulated blood for serum |
| • If a patient is a young child and 20ml of blood cannot be drawn, 2–3ml can be collected from each of the samples (6–9ml in total) |
| • Blood with EDTA must be immediately centrifuged at 3000rpm for 10min at 4°C followed by plasma collection in a clean tube, taking care so as not to absorb any red cells. After labelling 5 tubes with the patient's name, the date, and an indication showing EDTA plasma, plasma collected from these tubes should be distributed, freezing them immediately and keeping them at a temperature of −80°C |
| • Once plasma was drawn, store the cellular pellet frozen at −80°C for future DNA collection |
| • Coagulated blood for 1h at room temperature should be centrifuged at 3000rpm for 10min at 4°C and the serum should be immediately separated, taking care so as not to absorb any red cells in the clean tube. Following labelling of 5 tubes with the patient's name, the date, and an indication showing serum, serum collected from these tubes should be distributed, freezing samples immediately after and maintaining them at −80°C |
EDTA: ethylenediaminetetraacetic acid.
Genetic diagnosis of complement genes is generally recommended as it allows for individualised estimates of prognosis and risk of disease recurrence. Genetic tests are essential for patients who may be eligible for RT.
A Working Group coordinating and providing online counselling on complement tests for aHUS patients has been set up in order to collect data and conduct further trials.
Graft samples from renal transplant patients with aHUS should be collected for future studies.
Protocol for collection and shipping of samples for determination of ADAMTS13 activity.
| • Collect blood by venopuncture in two 5ml citrate tubes |
| • Label tubes with patient's name and last name, date, and time of collection |
| • Centrifuge tubes for 7min at 3500rpm |
| • Transfer supernatant (plasma) to Eppendorf's tubes for aliquots. Avoid transferring part of the precipitate (results may be altered). Samples must not be frozen before this step |
| • Label new tubes with the same information |
| • Freeze tubes at −20°C and send them to a reference laboratory. Also send a 5ml EDTA tube (refrigerated) |
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; EDTA: ethylenediaminetetraacetic acid.
Please cite this article as: Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. Actualización en síndrome hemolítico urémico atípico: diagnóstico y tratamiento. Documento de consenso. Nefrologia. 2015;35:421–447.



