





Renal fibrosis is the main cause of the development of diabetic kidney disease (DKD). ACSL1 plays an important role in colon cancer and liver fibrosis.
MethodsWe screened ACSL1 by proteomics analysis and then verified the expression of ACSL1 in the urine of diabetic nephropathy patients by WB and ELISA. Then, a total of 12db/m and db/db mice were used to verify the association between renal fibrosis and ACSL1. Periodic acid-Schiff (PAS) staining, Masson staining, and immunostaining were performed for histological studies. The relationship between ACSL1 and renal fibrosis was studied by knocking down ACSL1 in cell experiments.
ResultsThe expression of ACSL1 was significantly increased in the exfoliated urine cells and urine supernatant of diabetic nephropathy patients and was closely related to renal function. In addition, the expression of ACSL1 was significantly increased in the renal tissues of db/db mice with fibrosis. Knocking down ACSL1 in HK-2 cells was shown to reverse renal fibrosis induced by high glucose.
ConclusionsWe found a potential therapeutic target for preventing or ameliorating the progression of DKD fibrosis. Reducing ACSL1 expression may be a new strategy for the treatment of renal fibrosis caused by DKD, which provides an experimental theoretical basis for new drug research.
La fibrosis renal es la causa principal de desarrollo de nefropatía diabética (ND). ACSL1 juega un papel importante en el cáncer de colon y la fibrosis hepática.
MétodosCribamos ACSL1 mediante análisis proteómico, verificando seguidamente la expresión de ACSL1 en la orina de los pacientes con nefropatía diabética mediante WB y ELISA. A continuación, utilizamos un total de 12 ratones db/m y db/db para verificar la asociación entre fibrosis renal y ACSL1. Se realizaron tinciones PAS (Periodic acid-Schiff), de Masson e inmunotinción para los estudios histológicos. Estudiamos la relación entre ACSL1 y fibrosis renal aplicando la técnica de knockdown a ACSL1 en los experimentos celulares.
ResultadosLa expresión de ACSL1 se incrementó significativamente en las células de orina exfoliada y el sobrenadante de orina de los pacientes con nefropatía diabética y estuvo estrechamente relacionada con la función renal. Además, la expresión de ACSL1 se incrementó significativamente en los tejidos renales de los ratones db/db con fibrosis. La realización de knockdown a ACSL1 en las células HK-2 reflejó una reversión de la fibrosis renal inducida por la alta tasa de glucosa.
ConclusionesEncontramos un objetivo terapéutico potencial para prevenir o mejorar la progresión de la fibrosis en la ND. Reducir la expresión de ACSL1 puede suponer una nueva estrategia para el tratamiento de la fibrosis renal causada por la ND, lo cual aporta una base teórica experimental para la investigación de un nuevo fármaco.
Diabetic kidney disease (DKD) is the main cause of end-stage renal disease.1 The mechanisms leading to DKD are complex, and tubulointerstitial fibrosis is a common pathway for most renal injuries2 and one of the main pathological features of chronic kidney disease.3 Kidney fibroblasts play an important role in maintaining the homeostasis of the interstitial matrix and adjacent tissues under physiological conditions. Many cells are involved in the process of renal fibrosis. The main cause of tubulointerstitial fibrosis is the activation and expansion of fibroblasts, the production and deposition of a large number of extracellular matrix (ECM) components, and changes in both renal tubules and microvessels.4,5 Continued exposure to a high glucose (HG) environment induces epithelial-mesenchymal transition (EMT) in renal tubular epithelial cells, which in turn leads to interstitial fibrosis6 and the accumulation of extracellular matrix proteins such as collagen in the mesangial interstitium. The accumulation of protein and fibronectin leads to tubulointerstitial fibrosis.7,8 At present, the control of fibrosis development involves preventing, alleviating, and even reversing DKD by addressing the interaction between hemodynamic and metabolic pathways9 and controlling blood sugar, blood pressure and blood lipids using the renin–angiotensin–aldosterone system. The main drugs that block the action and can treat dyslipidemia are also effective agents for preventing diabetic nephropathy.10 However, many patients develop DKD despite the control of blood sugar, lipids, and blood pressure.11,12 By 2040, the prevalence of diabetes is expected to exceed 600 million, of which more than 20 million will develop DKD.13 Therefore, it is urgent to explore potential therapeutic targets for DKD to slow down the occurrence and development of DKD.
Acyl-CoA synthase long-chain family member 1 (long-chain-fatty-acid–CoA ligase 1, ACSL1) is an enzyme in the initial step of fatty acid β-oxidation and plays a key role in lipid biosynthesis and fatty acid degradation. Fatty acids are essential nutrients for the human body and can be obtained from diet or from de novo synthesis and breakdown of cellular triacylglycerols (TAGs) and phospholipids. As an important part of the body, these factors are also involved in energy metabolism and cell signaling pathways to maintain physiological functions. Long-chain acyl-CoA synthase (ACSL) activates fatty acids for intracellular metabolism and is also involved in the regulation of uptake.14 However, dysregulation of fatty acid metabolism leads to excessive lipid biosynthesis and deposition, which ultimately predisposes the body to metabolic disorders.15 Some studies have shown that ACSL1 is directly involved in lipid deposition in the kidney.16 In addition, in a study of colorectal cancer, it was found that ACSL1 could cause the transition of epithelial cells to mesenchymal cells through the AMPK signaling pathway. At the same time, the markers N-cadherin and Slug were upregulated, suggesting that ACSL1 could serve as a direct therapeutic target for colorectal cancer.17 However, ACSL1 has been poorly studied in diabetic kidney disease.
In addition, the use of urine proteomics and bioinformatics can provide insight into diabetic kidney disease,18 and it is also possible to predict the occurrence and development of DKD by using biomarkers in urine.19,20 GSK3b in exfoliated cells may serve as a novel biomarker for predicting the progression of diabetic kidney disease.21 Urine represents an infinite source of patient-specific kidney cells that can be obtained noninvasively. Podocytes and proximal tubule cells in urine have been used to investigate disease mechanisms and screen for novel drug therapies for various human kidney diseases.22
In the present study, we found that ACSL1 was significantly upregulated in the urine and kidney tissue of diabetic kidney disease patients and db/db mice. Knockdown of ACSL1 under HG conditions significantly alleviated renal fibrosis. Therefore, ACSL1 may be used as a therapeutic target to prevent or ameliorate the progression of DKD fibrosis.
Experimental procedureClinical studiesThis study was conducted at the Affiliated Hospital of Xuzhou Medical University from March to June 2022. A total of 84 urine samples were collected, including 20 urine samples from healthy people at the Health Management Center of the Affiliated Hospital of Xuzhou Medical University; urine samples were collected from 30 diabetic patients and 34 diabetic nephropathy patients in the Department of Endocrinology. Each individual's basic information and clinical parameters were recorded, including sex, age, medication, diabetes duration, blood pressure, blood sugar level, serum creatinine level, urine analysis data, UACR and eGFR. All patients provided informed consent.
Diagnostic criteria present for DKD are usually based on persistence albuminuria and/or eGFR levels decreased, while other sources were excluded a clinical diagnosis was made of induced chronic kidney disease (CKD). In defining diabetes as kidney etiology of lesions and exclusion of other causes of CKD in the case of to less than one of the following can be diagnosed as DKD: (1) eliminate interference factors at least 2 out of 3 tests within 3–6 months UACR≥30mg/g or urinary albumin excretion rate ≥30mg/24h (≥20μg/min); (2) eGFR<60mL/(min 1.73m2) lasted 3 times more than a month; (3) renal biopsy was consistent with the pathological changes of DKD.23
Urine samplesA 10mL urine sample from the first morning urine stream was collected. Urine samples were centrifuged at 3000×g for 15min and stored at −80°C. The supernatant was divided into 1.5mL portions and immediately stored at −80°C. The urine samples were centrifuged at 10,000×g for 15min, and the exfoliated cells were retained and immediately stored at −80°C.
ELISAA specific ELISA kit was used to detect the expression of ACSL1 in the urine of healthy subjects, diabetic patients and diabetic kidney disease patients.
AnimalsTwelve male C57BLKS/J mice with a Lep db/Lep db (db/db) (7 weeks old, 30–35g) background and nondiabetic control Lep db/m (db/m) littermate mice (7 weeks old, 15–20g) were purchased from the Model Animal Research Center of Nanjing University (Jiangsu, China). All animals were housed in a barrier environment and provided food and water ad libitum before and during the experiments. All experimental procedures were approved by the Animal Ethics Committee of Xuzhou Medical University. Some db/m and db/db mice were fed until they were 24 weeks old, and blood, urine and kidney tissues were collected to observe the expression of ACSL1 in the kidneys of diabetic nephropathy mice.
Detection of renal function and biochemical parametersRenal function was assessed through the measurement of blood urea nitrogen (BUN), blood creatinine (Cr) and urinary albumin levels. BUN and Cr were determined using assay kits from Jiancheng (Nanjing, China). Serum levels of fasting blood glucose (FBG) were measured by test strips from LifeScan (CA, USA). The kidney index (mg/g) was calculated as the ratio of the weight of the two kidneys to the body weight of the mouse.
Cell culture and transfectionThe human proximal renal tubular epithelial cell line HK-2 was a gift from Nanjing University. The cells were cultured in DMEM containing 5.56mmol/L glucose (normal glucose, NG) supplemented with 10% fetal bovine serum (Invitrogen, Grand Island, NY) and 1% penicillin–streptomycin in a humidified incubator at 37°C with 5% CO2. To induce EMT, the cells were cultured in medium containing 60mmol/L glucose for 72h. Glucose (5.56mmol/L) and mannitol were used for osmotic control (MA).
For ACSL1-shRNA experiments, HK-2 cells were stimulated with HG and transfected with the control lentivirus vector (HG+NC) or ACSL1+shRNA lentivirus vector (HG+ACSL1-shRNA) for 72h. HK-2 cells were cultured in HG without other treatments (HG) as a control.
Histopathological analysisKidney tissues were fixed in 10% phosphate buffered formalin and embedded in paraffin. Paraffin sections (2–3μm) were stained with periodic acid-Schiff (PAS) and Masson's trichrome.
Histology and immunohistochemistry analysisThe kidney samples were fixed in 4% paraformaldehyde and embedded in paraffin. Sections with a thickness of 5μm were cut perpendicular to the long axis of the kidney for immunohistochemistry and morphometric analysis.
Western blot analysesUrinary exfoliated cells were lysed with ice-cold RIPA buffer, and the insoluble materials were removed. The proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide (SDS) gel electrophoresis, transferred onto nitrocellulose (NC) membranes and blocked with 3% BSA for 2h at RT. The membranes were incubated with primary antibodies at 4°C overnight and then with horseradish peroxidase-conjugated secondary antibodies for 1h. The primary antibody used was anti-ACSL1 (Affinity). The intensities of the bands were quantified using an Odyssey® Sa system (LICOR, USA). Protein signals were quantified by optical density ratios using GAPDH expression as a control.
Statistical analysisStatistical analyses were performed using SPSS statistics 20.0 software, and all experimental data are expressed as the mean±SEM. Comparisons among groups were performed using one-way analysis of variance (ANOVA). Differences between two groups were assessed with Student's t test. Differences were considered significant if p<0.05.
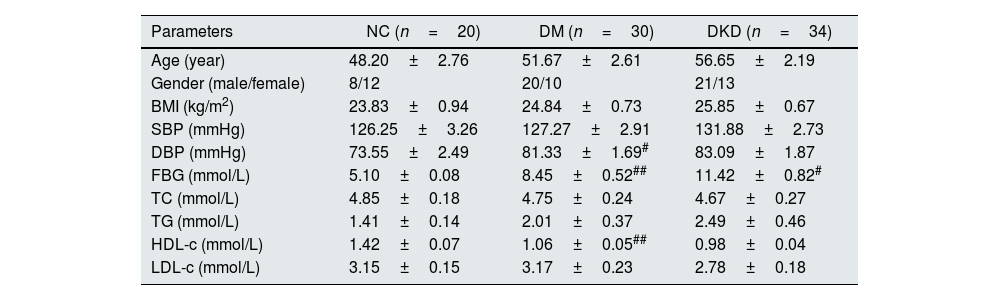
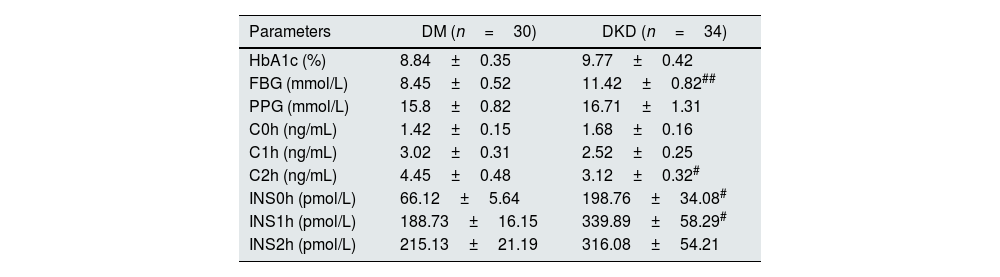
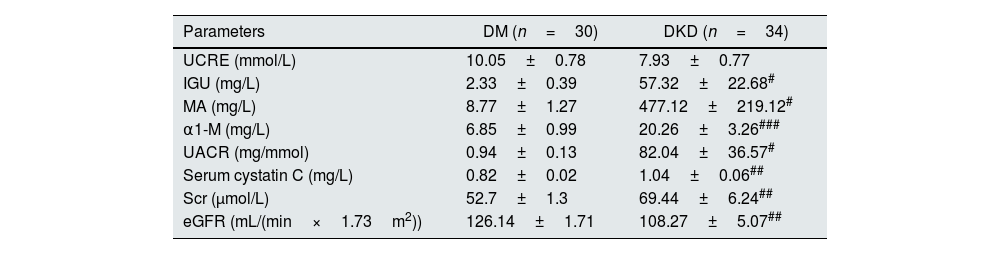
ResultsBasic clinical data and biochemical indicatorsFirst, the basic clinical information of the subjects was compared. The results showed that there were no significant differences in age, sex, body mass index (BMI), low-density lipoprotein cholesterol (LDL-C), triglycerides (TGs), or total cholesterol (TC) among the subjects. However, there were significant differences in fasting blood glucose, diastolic blood pressure, and high-density lipoprotein cholesterol (HDL-C) in DM patients compared with controls (p<0.05, Table 1). Compared with DM patients, there were also significant differences in fasting blood glucose, C2h, INS0h, and INS1h in DKD patients (p<0.05, Table 2). In addition, in terms of clinical renal function indicators, there were significant differences in IGU, MA, α1-M, UACR, blood Cys C, Scr and eGFR in DKD patients compared with DM patients (p<0.05, Table 3).
General data for each group.
| Parameters | NC (n=20) | DM (n=30) | DKD (n=34) |
|---|---|---|---|
| Age (year) | 48.20±2.76 | 51.67±2.61 | 56.65±2.19 |
| Gender (male/female) | 8/12 | 20/10 | 21/13 |
| BMI (kg/m2) | 23.83±0.94 | 24.84±0.73 | 25.85±0.67 |
| SBP (mmHg) | 126.25±3.26 | 127.27±2.91 | 131.88±2.73 |
| DBP (mmHg) | 73.55±2.49 | 81.33±1.69# | 83.09±1.87 |
| FBG (mmol/L) | 5.10±0.08 | 8.45±0.52## | 11.42±0.82# |
| TC (mmol/L) | 4.85±0.18 | 4.75±0.24 | 4.67±0.27 |
| TG (mmol/L) | 1.41±0.14 | 2.01±0.37 | 2.49±0.46 |
| HDL-c (mmol/L) | 1.42±0.07 | 1.06±0.05## | 0.98±0.04 |
| LDL-c (mmol/L) | 3.15±0.15 | 3.17±0.23 | 2.78±0.18 |
BMI: body mass index; SBP: systolic blood pressure; DBP: diastolic blood pressure; FBG: fasting blood glucose; TC: total cholesterol; TG: triglycerides; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; NC: nitrocellulose; DM: diabetes mellitus; DKD: diabetic kidney disease; #p<0.05, ##p<0.001, compared with NC.
Basic clinical indicators of the DM group and DKD group.
| Parameters | DM (n=30) | DKD (n=34) |
|---|---|---|
| HbA1c (%) | 8.84±0.35 | 9.77±0.42 |
| FBG (mmol/L) | 8.45±0.52 | 11.42±0.82## |
| PPG (mmol/L) | 15.8±0.82 | 16.71±1.31 |
| C0h (ng/mL) | 1.42±0.15 | 1.68±0.16 |
| C1h (ng/mL) | 3.02±0.31 | 2.52±0.25 |
| C2h (ng/mL) | 4.45±0.48 | 3.12±0.32# |
| INS0h (pmol/L) | 66.12±5.64 | 198.76±34.08# |
| INS1h (pmol/L) | 188.73±16.15 | 339.89±58.29# |
| INS2h (pmol/L) | 215.13±21.19 | 316.08±54.21 |
HbA1c: hemoglobin A 1c; FBG: fasting blood glucose; PPG: postprandial blood glucose; C0h: C-peptide level at 0h postprandial; C1h: C-Peptide level at 1h postprandial; C2h: C-peptide level at 2h postprandial; INS0h: insulin level at 0h postprandial; INS1h: insulin level at 1h postprandial; INS2h: insulin level at 2h postprandial; DM: diabetes mellitus; DKD: diabetic kidney disease.
#p<0.05, ##p<0.01, compared with DM patients.
Clinical renal function indexes of the DM group and DKD group.
| Parameters | DM (n=30) | DKD (n=34) |
|---|---|---|
| UCRE (mmol/L) | 10.05±0.78 | 7.93±0.77 |
| IGU (mg/L) | 2.33±0.39 | 57.32±22.68# |
| MA (mg/L) | 8.77±1.27 | 477.12±219.12# |
| α1-M (mg/L) | 6.85±0.99 | 20.26±3.26### |
| UACR (mg/mmol) | 0.94±0.13 | 82.04±36.57# |
| Serum cystatin C (mg/L) | 0.82±0.02 | 1.04±0.06## |
| Scr (μmol/L) | 52.7±1.3 | 69.44±6.24## |
| eGFR (mL/(min×1.73m2)) | 126.14±1.71 | 108.27±5.07## |
UCRE: urine creatinine; IGU: urinary immunoglobulin G; MA: microalbumin; α1-M: urinary α1-microglobulin; UACR: microalbumin/urine creatinine; Scr: serum creatinine; eGFR: estimated glomerular filtration rate; DM: diabetes mellitus; DKD: diabetic kidney disease; #p<0.05, ##p<0.01, ###p<0.001, compared with DM patients.
Subsequently, the expression levels of ACSL1 in the urine exfoliated cells of the different test groups were detected by WB analysis. Compared with the NC group and the DM group, the expression level of ACSL1 in the urinary exfoliated cells in the DKD group was significantly increased (Fig. 1A). The expression levels of ACSL1 in the urine supernatants of the different test groups were detected by ELISA. Compared with the NC group and the DM group, the expression level of ACSL1 in the urinary supernatant of the DKD group was also significantly increased (Fig. 1B). More notably, the upregulation of ACSL1 in the urine of DKD patients was positively correlated with UACR (Fig. 1C), immunoglobulin G (Fig. 1D), and cystatin C (Fig. 1E) and negatively correlated with eGFR (Fig. 1F). These results suggest that ACSL1 may be involved in the pathogenesis of DKD.
 WB analysis demonstrated that the expression level of ACSL1 was significantly increased in the urinary exfoliated cells of DKD patients. (B) ELISA showed that the expression level of ACSL1 was significantly increased in the urine supernatant of DKD patients. (C) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and UACR. (D) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and IGU. (E) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and serum cystatin C. (F) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and eGFR. *p<0.05, compared with NC; #p<0.05, compared with DM patients. The expression of ACSL1 in urine supernatants from NC (20 healthy person), DM (30 diabetes mellitus) and DKD (34 diabetic kidney disease) by ELISA.")
The expression of ACSL1 is increased in the urine of DKD patients, and there is a correlation with renal function in DKD patients. (A) WB analysis demonstrated that the expression level of ACSL1 was significantly increased in the urinary exfoliated cells of DKD patients. (B) ELISA showed that the expression level of ACSL1 was significantly increased in the urine supernatant of DKD patients. (C) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and UACR. (D) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and IGU. (E) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and serum cystatin C. (F) Spearman correlation analysis was used to detect the correlation between urine supernatant ACSL1 and eGFR. *p<0.05, compared with NC; #p<0.05, compared with DM patients. The expression of ACSL1 in urine supernatants from NC (20 healthy person), DM (30 diabetes mellitus) and DKD (34 diabetic kidney disease) by ELISA.
To investigate the effect of HG on ACSL1 expression in vivo, we constructed a db/db mouse model and conducted experiments, urinary microalbumin to creatinine (mAlb/Cr) levels and fasting blood glucose (FBG) levels in db/db mice were markedly elevated compared with those in db/m mice. Compared with db/m mice, PAS staining showed that renal tubular dilatation was more obvious in db/db mice, indicating renal tubular injury (Fig. 2A). Compared with db/m mice, the distribution of Masson-positive areas was significantly increased in db/db mice, showing collagen accumulation in the renal cortex (Fig. 2B). The results showed that renal function was decreased in diabetic mice. In addition, the expression of total ACSL1 protein was significantly increased in db/db mice (Fig. 2C and D), and immunohistochemistry showed that this expression was more pronounced around renal tubules (Fig. 2E). These results suggest that ACSL1 may be involved in the occurrence of diabetic kidney disease in mice.
 Periodic acid-Schiff staining of the renal cortex sections of db/m and db/db mice. (B) Masson staining of the renal cortex sections of db/m and db/db mice. (C) WB results demonstrated the expression levels of total ACSL1 in mice. (D) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (E) The expression of ACSL1 in mice was assessed by immunohistochemistry. Data information: all values are shown as the means±SEMs of 6–8 rats. *P<0.05, mean db/db mice compared with db/m mice.")
ACSL1 expression was increased in db/db mouse models. (A) Periodic acid-Schiff staining of the renal cortex sections of db/m and db/db mice. (B) Masson staining of the renal cortex sections of db/m and db/db mice. (C) WB results demonstrated the expression levels of total ACSL1 in mice. (D) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (E) The expression of ACSL1 in mice was assessed by immunohistochemistry. Data information: all values are shown as the means±SEMs of 6–8 rats. *P<0.05, mean db/db mice compared with db/m mice.
Then, we validated these changes in vitro. HK-2 cells were treated with HG medium (60mmol/L d-glucose) for 72h. The results showed that HG promoted the expression of ACSL1 (Fig. 3A and B), and to eliminate the effect of HG-induced osmotic changes, mannitol was used as an osmotic control. In addition, HG inhibited the expression of E-cadherin and Zo-1 and increased the expression of α-SMA and Vimentin, which are mesenchymal proteins. These data suggest that HG induces EMT in HK-2 cells.
 WB analysis demonstrated the expression levels of ACSL1 in HK-2 cells under normal glucose (NG), high glucose (HG) or mannitol (MA) treatment for 72h. (B) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (C) WB analysis demonstrated the expression levels of EMT-related proteins in HK-2 cells under normal glucose (NG), high glucose (HG) or mannitol (MA) conditions for 72h. (D) Quantitation of the arbitrary ratios of EMT-related proteins to GAPDH. Data information: all values are shown as the means±SEMs of at least three independent experiments. *P<0.05; **P<0.01, mean HG compared with NG.")
Effect of HG on ACSL1 protein levels and EMT-related proteins. (A) WB analysis demonstrated the expression levels of ACSL1 in HK-2 cells under normal glucose (NG), high glucose (HG) or mannitol (MA) treatment for 72h. (B) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (C) WB analysis demonstrated the expression levels of EMT-related proteins in HK-2 cells under normal glucose (NG), high glucose (HG) or mannitol (MA) conditions for 72h. (D) Quantitation of the arbitrary ratios of EMT-related proteins to GAPDH. Data information: all values are shown as the means±SEMs of at least three independent experiments. *P<0.05; **P<0.01, mean HG compared with NG.
To further clarify the role of ACSL1 in EMT during DKD, we reduced the protein expression of ACSL1 in HG-induced HK-2 cells (Fig. 4A and B) and found that the expression of epithelial cell markers (E-cadherin and ZO-1) was significantly upregulated. The expression of mesenchymal cell markers (vimentin and α-SMA) was downregulated (Fig. 4C and D). Taken together, these results suggest that ACSL1 knockout can effectively improve EMT during renal fibrosis induced by diabetic kidney disease.
 WB analysis demonstrated the expression levels of ACSL1 in HK-2 cells. (B) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (C) WB analysis demonstrated the expression levels of EMT-related proteins in HK-2 cells. (D) Quantitation of the arbitrary ratios of EMT-related proteins to GAPDH. HG: cells treated with 60mmol/L glucose; HG+NC: cells treated with HG and transfected with NC lentivirus; and HG+ACSL1-shRNA: cells treated with HG and transfected with ACSL1-shRNA lentivirus. Data information: all values are shown as the means±SEMs of at least three independent experiments. *P<0.05, **P<0.01, mean HG+ACSL1-shRNA compared with HG.")
Effects of ACSL1 knockdown on renal fibrosis and EMT in HK-2 cells. (A) WB analysis demonstrated the expression levels of ACSL1 in HK-2 cells. (B) Quantitation of the arbitrary ratios of ACSL1 to GAPDH. (C) WB analysis demonstrated the expression levels of EMT-related proteins in HK-2 cells. (D) Quantitation of the arbitrary ratios of EMT-related proteins to GAPDH. HG: cells treated with 60mmol/L glucose; HG+NC: cells treated with HG and transfected with NC lentivirus; and HG+ACSL1-shRNA: cells treated with HG and transfected with ACSL1-shRNA lentivirus. Data information: all values are shown as the means±SEMs of at least three independent experiments. *P<0.05, **P<0.01, mean HG+ACSL1-shRNA compared with HG.
Diabetic kidney disease (DKD) is the main cause of end-stage renal disease,1 which is one of the most common and serious complications of diabetes mellitus (DM).24 There are many factors that cause diabetic nephropathy, and its pathogenesis is complicated. At present, its pathogenesis is not fully understood, which leads to poor treatment outcomes. Standard treatment mainly controls blood glucose, blood pressure and blood lipids. However, despite the control of blood glucose, blood pressure and blood lipids, many patients still develop DKD.11 In recent years, an increasing number of studies have attempted to find therapeutic targets for DKD to delay or control the occurrence and development of DKD.
Urine is produced by glomerular filtration and reabsorption in the renal tubules. The exfoliated renal cells in urine contain biological information and can also reflect the degree of renal tissue damage.25 Urine specimens are noninvasive and readily available, and the use of urine proteomics and bioinformatics analyses provides insights into diabetic kidney disease.18 In the present study, we measured the expression of ACSL1 in urine exfoliated cells by WB and detected the expression of ACSL1 in urine supernatants by ELISA. Compared with that in healthy and diabetic patients, the expression of ACSL1 in the urine of DKD patients was significantly higher (Fig. 1A and B), which was closely related to renal function in DKD (Fig. 1D–F). Therefore, we hypothesize that ACSL1 may be involved in the occurrence and development of DKD. However, the small sample size was a limitation of the study. In this study, we only enrolled 84 patients based on our inclusion criteria for healthy individuals, diabetic patients, and diabetic nephropathy, so we did not have sufficient statistical power to detect the effect of ACSL1 on some clinical indicators.
Long-chain acyl-CoA synthase (ACSL) activates fatty acids for intracellular metabolism and participates in the regulation of uptake.14 However, dysregulation of fatty acid metabolism leads to excessive lipid biosynthesis and deposition, which ultimately makes the body prone to metabolic disorders.15 ACSL1 also plays a very important role in regulating TG synthesis in the liver.26 Studies have shown that microRNA-34A and miR-34C target ACSL1 and reduce the expression of ACSL1, thus reducing the expression of α-SMA, type I collagen and desmin and alleviating liver fibrosis,27,28 indicating that the overexpression of ACSL1 promotes liver fibrosis. Therefore, we considered whether the overexpression of ACSL1 promoted the occurrence and development of DKD-induced renal fibrosis.
Renal interstitial fibrosis is an important pathological basis for the development of diabetic nephropathy. Compared with glomerulosclerosis, renal interstitial fibrosis can more accurately reflect the degree of kidney damage, which leads to renal failure. Although the mechanism of renal fibrosis is still unclear, increasing evidence indicates that epithelial-mesenchymal transition (EMT) plays an important role in the process of renal fibrosis.29 EMT can occur under various conditions, such as in an animal model of renal fibrosis.30 During EMT, epithelial cells lose characteristic markers, such as E-cadherin and cytokeratin, and gain mesenchymal cell-specific morphological and molecular markers, including α-smooth muscle actin (α-SMA), fibronectin (FN), and type I collagen.31 Epithelial cells with EMT do not directly generate interstitial myofibroblasts. Instead, these cells release signals that promote the transformation of fibroblasts into myofibroblasts. EMT describes a complex phenotypic change in epithelial cells.32 Therefore, we constructed a db/db mouse model and used PAS and Masson staining to show that the kidneys of db/db mice showed obvious renal tubular injury and fibrosis compared with those of db/m mice (Fig. 2A and B). The expression of ACSL1 was upregulated in db/db mouse renal tissues, as shown by WB, and the expression of ACSL1 around renal tubules was also significantly upregulated, as shown by immunohistochemistry (Fig. 2C–E). These results suggest that ACSL1 may play an important role in DKD-induced renal fibrosis.
Studies have shown that DKD is closely related to fibrosis in renal tubular epithelial cells.33 Epithelial cells, which are located in the outer layer of the renal tubule, are responsible for reabsorbing glucose, amino acids and other substances in urine.34 Early studies have shown that exposure to high glucose or albumin levels can induce fibrosis in renal tubule epithelial cells. This effect is closely related to the increased expression of McP-1, PAI-1 and TGF-β1 due to ROS production induced by hyperglycemia.35 In addition, fibrosis in renal tubular epithelial cells is closely related to proteinuria.36 Hyperglycemia can also cause renal tubular epithelial cells to lose polarity and acquire migration and invasion characteristics,37 leading to increased expression of fibronectin and α-smooth muscle actin (α-SMA) and decreased expression of E-cadherin, which ultimately lead to fibrosis. We stimulated HK-2 cells with an HG concentration of 60mmol/L for 72h, and Western blotting showed that ACSL1 expression was increased by HG compared with NG (Fig. 3A and B). Moreover, EMT-related proteins and the epithelial cell markers ZO-1 and E-cadherin, were downregulated, and the mesenchymal cell markers vimentin and α-SMA were upregulated (Fig. 3C and D), which was consistent with other studies. To further verify that these observed changes were the result of HG itself and not of HG-induced osmotic pressure changes, an osmotic pressure control was included in our study, and the results excluded the possibility of in vitro osmotic pressure changes.
Subsequently, we hypothesized that decreased ACSL1 expression could inhibit renal fibrosis and contribute to the treatment of DKD-induced renal fibrosis. We conducted further studies of HK-2 cells to reveal the role of ACSL1 in EMT during DKD. The recovery of EMT-related protein expression in cells treated with HG and ACSL1-shRNA confirmed our previous hypothesis (Fig. 4C and D). Therefore, our data suggest that ACSL1 may be a new therapeutic target for DKD-associated renal fibrosis.
In conclusion, we found a potential therapeutic target for preventing or ameliorating the progression of DKD-associated fibrosis. We found that the expression of ACSL1 was significantly increased in the urine of patients with DKD. Then, by constructing a db/db mouse model, we found that severe renal fibrosis occurred in mice, and the expression of ACSL1 was significantly increased in kidney tissue. ACSL1 expression was also increased in HK-2 cells in response to HG stimulation. In addition, gene-specific downregulation of ACSL1 expression could reverse HG-induced EMT. Therefore, targeting ACSL1 may be a promising therapeutic strategy for the treatment of DKD, renal fibrosis and other metabolism-related fibrotic diseases. In addition, these results may pave the way for large-scale, in-depth studies of ACSL1 as a therapeutic target for DKD and renal fibrosis.
ConclusionIn summary, ACSL1 may become a target for research for the treatment of diabetic nephropathy fibrosis, and our results provide an effective and accurate experimental basis for the research of new drugs.
Availability of data and materialsThe data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributionsParticipated in research design: Xueyan Zhou, Zhonghui Cao and Xiao Gao.
Conducted experiments: Zhonghui Cao, Xiao Gao, Jing Meng, Xiaoli Guo, Jiahao Xu and Junchao Cui.
Contributed new reagents or analytic tools: Jing Meng, Xiaoli Guo, Jiahao Xu, Junchao Cui.
Performed data analysis: Zhonghui Cao, Xiao Gao and Xueyan Zhou.
Wrote or contributed to the writing of the manuscript: Zhonghui Cao, Xiao Gao and Xueyan Zhou.
FundingThis study was supported by the Natural Science Foundation of China [No. 82173883]; the Science and Technology Foundation of Xuzhou [No. KC21010, KC22469]; the Natural Science Foundation of the Jiangsu Higher Education Institutions of China [No. 18KA350002]; the Natural Science Foundation of Jiangsu Province [No. BK20181470]; the Provincial Commission of Health and Family Planning in Jiangsu Province [No. H2017079] and the Science and Technology Planning Project of Jiangsu Province [No. BE2019636].
Conflict of interestsThe authors declare that they have no competing interests.
We thank all volunteers in this study for their cooperation, and we thank the physicians from the Department of Endocrinology, the Affiliated Hospital of Xuzhou Medical University for their support.



