

Desde la aparición en 1989 de la eritropoyetina humana recombinante (rHuEPO), muchos pacientes se han beneficiado de este tratamiento. Durante este tiempo hemos aprendido a utilizar dosis menores, manejar adecuadamente la ferroterapia acompañante, establecer los objetivos de hemoglobina, corregir problemas asociados y evitar efectos adversos con un uso juicioso de estas moléculas combinadas con distintas pautas de ferroterapia. Hasta 2008 se han desarrollado modificaciones de la estructura de la rHuEPO para aumentar su vida media y mejorar su eficacia y seguridad. En este artículo se revisa la evolución, el desarrollo histórico y los problemas presentados durante los últimos 20 años, como los valores objetivos o la aplasia pura de células rojas. El presente artículo sirve de introducción histórica al resto de artículos del presente suplemento monográfico.
Since the appearance of recombinant human erythropoietin (rhEPO) in 1989, many patients have benefited from this treatment. During this time, we have learned to use lower doses, adequately manage the associated iron therapy, establish haemoglobin targets, correct associated problems and avoid adverse effects with the judicious use of these molecules combined with distinct iron therapy regimens. Until 2008, the structure of rhEPO was modified to increase its half-life and improve its safety and efficacy. The present article revises the historical development of these agents and the problems encountered in the last 20 years, such as target values and pure red cell aplasia. The present article serves as a historical introduction to the remaining articles in the present supplement.
ERiTRoPoyETiNAS hUMANAS RECoMBiNANTES
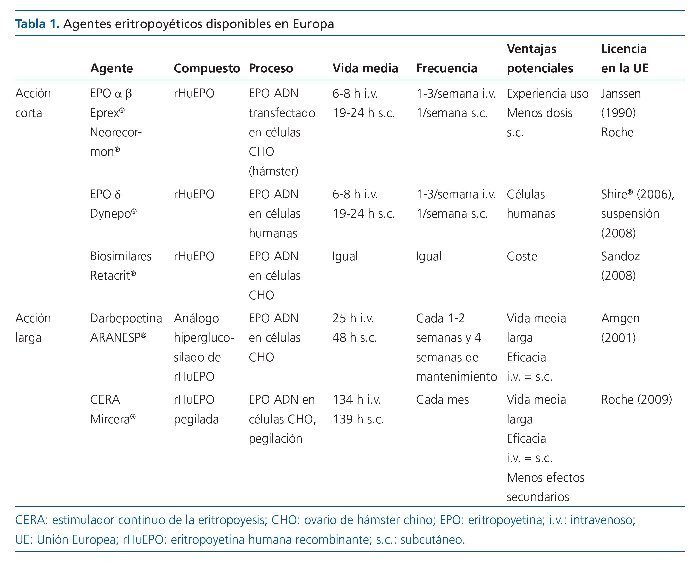
La eritropoyetina humana recombinante (rHuEPO) es una glucoproteína de 30,4 kD, con 165 aminoácidos y 3 grupos N-glicanos con diferentes isómeros. La eritropoyetina (EPO) b tiene mayor proporción de grupos tetrasiálicos (el 46% frente al 19%), lo que le confiere un mayor peso molecular y una vida media discretamente mayor. Estas pequeñas diferencias entre rHuEPO a y b carecen de relevancia clínica, por lo que se considera que todas las rHuEPO de acción corta tienen la misma pauta de uso, eficacia y seguridad y son equivalentes1,2.
Se sabe que la vía subcutánea (s.c.) es más eficiente (menores dosis para igual respuesta) y que la administración en intervalos prolongados asocia una dosis mayor (dose penalty). Sin embargo, la utilización rutinaria está mediada por factores locales de experiencia en el centro o factores prácticos del perfil de paciente3. En España, lo habitual es usar la vía intravenosa (i.v.) y una pauta coincidente con 2 o 3 sesiones de hemodiálisis a la semana. En el caso de los pacientes en diálisis peritoneal o con enfermedad renal crónica (ERC) avanzada, se usa la vía s.c. con intervalos semanales o incluso quincenales en fases de mantenimiento.
La rHuEPO d (Shire®) aportaba la ventaja conceptual de ser producida en líneas celulares humanas. Sin embargo, no pudo demostrar que ello tuviera impacto en resultados clínicos antes de que se interrumpiera su comercialización en 2009. La rHuEPO z (Eporatio® Ratiopharm; Retacrit® Hospira) fue aprobada en la Unión Europea (UE) por la Agencia Europea del Medicamento en 2009. Es otra variante producida en líneas celulares de hámster, con pequeñas diferencias en estructura frente a rHuEPO a y b, lo que no se considera un biosimilar y no aporta diferencias en resultados clínicos.
Simultáneamente se desarrollaron rHuEPO biosimilares de rHuEPO a comercializadas en la UE en 2008. En España está disponible Binocrit® (Sandoz, Austria). Se trata de productos recombinantes similares a la molécula original, elaborados por un fabricante diferente, en nuevas líneas celulares, con nuevos procesos, por lo que no son exactamente idénticos. Los biosimilares tienen un proceso regulatorio específico, que controla los procesos de producción y exige estudios clínicos de no inferioridad con respecto a las moléculas originales. Se le atribuyen los mismos resultados (efecto grupo) que las rHuEPO originales de acción corta y han aportado escasa evidencia añadida, aparte de resultados controvertidos sobre coste-eficacia4. De hecho, la reciente Revisión Cochrane no encuentra diferencias de efectividad entre los distintos agentes comercializados que justifiquen el uso prioritario de ninguno ellos5. La regulación en el desarrollo y comercialización de biosimilares pretende garantizar la calidad del tratamiento y la seguridad de uso, y la norma es la prescripción por marca comercial y la prohibición de sustitución automática entre ellos. Todo ello para facilitar la trazabilidad del tratamiento ante la aparición de eventuales efectos adversos y el seguimiento de protocolos específicos de farmacovigilancia6,7. Esto es especialmente relevante para el seguimiento de casos de aplasia pura de células rojas, una complicación poco frecuente (tasas de incidencia de 1 caso por cada 20.000 paciente-año de tratamiento con EPO a original), de la que ya se han descrito 2 casos en una serie de 337 pacientes tratados con Binocrit®8.
Progresivamente se han realizado varias modificaciones encaminadas a prolongar la vida media de las rHuEPO y a mejorar su tolerancia (tabla 1). En el año 2000 se comercializó en nuestro país darbepoetina, una molécula de EPO genéticamente modificada con un aumento en sus grupos glucosilados, que consigue una vida media 5 veces mayor y permite pautas s.c. quincenales e incluso mensuales en fase de mantenimiento en determinados pacientes estables9. A partir de 2009 estuvo disponible la EPO b pegilada (estimulador continuo de la eritropoyesis), con una vida media 20 veces mayor y con farmacocinética similar para ambas vías de administración (i.v. o s.c.), lo que permitía iniciar directamente el tratamiento con pauta mensual en fase de corrección. Este preparado de acción larga prometía mayor estabilidad en los valores de hemoglobina (Hb), menores ajustes de dosis, comodidad de administración, reducción relativa de dosis en pacientes con altos requerimientos y precio equivalente a los preparados de acción corta10. Sin embargo, la concurrencia de los biosimilares y de un problema en la comercialización coincidente con el lanzamiento, ha reducido mucho su uso en nuestro país.

Desde las fases iniciales de la comercialización de la primera rHuEPO, se aceptó tácitamente que no era preciso realizar grandes estudios controlados, bien diseñados y con eventos duros para definir los objetivos por grupos de pacientes, dosis y pautas. La base fisiopatológica era intachable, la tasa de respuesta muy elevada y la mejoría clínica inmediata con muy baja tasa de eventos adversos. Los estudios realizados en pacientes con ERC no en diálisis (CHOIR, TREAT, CREATE), no consiguieron demostrar que la normalización indiscriminada de los valores de Hb consiguiera una reducción de la mortalidad ni de la progresión de la ERC11. Es más, esta aproximación simplista al problema (objetivo común para todos y subir dosis hasta alcanzar el objetivo) conlleva un aumento del riesgo cardiovascular (CV) y ha supuesto una pérdida de oportunidad para obtener mejor evidencia12,13. Tras estos resultados, las guías de práctica clínica fijan el objetivo genérico en 10-12 g/dl de Hb y proponen individualizar el objetivo para cada paciente (según técnica de diálisis, edad, comorbilidad, etc.), aunque sin recomendaciones precisas basadas en estudios. Los análisis post-hoc demostraron finalmente que los eventos CV estaban más asociados a las dosis elevadas de los pacientes más resistentes (y más comórbidos) que a la concentración de Hb alcanzada per se14. Se postuló por los efectos pleiotrópicos de la EPO sobre la interacción endotelio-plaquetas13.
Por desgracia, el escenario actual del tratamiento convencional de la anemia con EPO muestra una carencia de estudios para resolver temas pendientes entre el desinterés de las compañías farmacéuticas y un manejo de la selección de proveedores basado en la continua guerra de ofertas.
Los nuevos productos en desarrollo pueden agruparse en 4 subgrupos: péptidos EPO miméticos, estabilizadores del HIF (factor inducible por hipoxia), reguladores de la hepcidina y otros fármacos en fases precoces de desarrollo, que se revisan en el próximo artículo del presente monográfico15.
Conflicto de intereses
J. Portolés manifiesta que ha participado como investigador en ensayos sobre la anemia patrocinados por Amgen, Roche y Takeda. A. Cases y A. Martínez Castelao manifiestan que han recibido honorarios de Roche, Amgen y Vifor por la realización de conferencias. J.L. Górriz ha participado en conferencias patrocinadas por Vifor Pharma. B. Quiroga, J.M. López, A.L. Martín de Francisco, M. Arias y P. Aljama no presentan conflicto de intereses.
CoNCEPToS CLAVE
• El desarrollo de agentes estimulantes de la eritropoyesis (ESA, del inglés erythropoiesis-stimulating agent) se ha basado en modificar aspectos de farmacocinética y farmacodinamia que faciliten su uso y seguridad.
• Las pautas y objetivos del tratamiento de la anemia de la ERC con ESA y ferroterapia están consolidados en las guías clínicas, pero es importante la individualización.
• El desarrollo del tratamiento de la anemia abre vías prometedoras más allá de la síntesis de variantes de rHuEPO y de la modificación de su farmacocinética y farmacodinamia.
Correspondencia:
josem.portoles@salud.madrid.org
* Relación de nombres del Grupo de Anemia de la SEN en el anexo.


