








In the last five years, the medical community was astonishingly surprised by the sequential large outcome trials that displayed the renal effects of sodium glucose co-transporter inhibitors (SGLT2Is) in type 2 diabetes mellitus (T2DM) patients with or without chronic kidney disease (CKD). This favorable effect was later disclosed in non-diabetic CKD patients. The EMPA-REG OUTCOME trial was the first trial that showed a reduction for the need for dialysis in patients suffering diabetic kidney disease (DKD) by 55%. This figure is double the score achieved by the angiotensin receptor blocker, Losartan, in RENAAL trial. The need for dialysis in DAPA-CKD trial was reduced in diabetic and non-diabetic CKD patients by 33%. The renal-specific composite outcome was reduced by 39% in EMPA-REG trial, 40% in CANVAS study, 47% in DECLARE-TIMI 58 study, 34% in CREDENCE trial, and 44% in DAPA-CKD trial. The greater surprise is the significant favorable effect of SGLT2Is on overall mortality in CKD patients with or without T2DM. Similar survival benefit was not previously encountered with any of the medications used in CKD patients with or without diabetes. In this review, we disclose the results of the DAPA-CKD trial, the CREDENCE trial and those of several cardiovascular outcome trials (CVOT) that used different SGLT2Is and showed that patients with lower eGFR levels may have greater benefit with respect to cardiovascular morbidity than patients with normal kidney function. In addition, we discuss the different mechanisms of action that explain the renal beneficial effects of SGLT2Is.
Durante los últimos cinco años la comunidad médica se ha visto sorprendida por los grandes ensayos de resultados secuenciales que mostraron los efectos renales de los inhibidores del cotransportador de sodio y glucosa (SGLT2I) en pacientes con diabetes mellitus de tipo 2 (DMT2), con o sin enfermedad renal crónica (ERC). Este efecto favorable se descubrió posteriormente en pacientes no diabéticos con ERC. El ensayo EMPA-REG OUTCOME fue el primero que mostró una disminución del 55% de la necesidad de diálisis en pacientes con enfermedad renal diabética (ERD). Esta cifra duplica la puntuación obtenida por el antagonista de los receptores de la angiotensina (losartán) en el ensayo RENAAL. La necesidad de diálisis en el ensayo DAPA-CKD se redujo en un 33% en los pacientes con ERC diabéticos y no diabéticos. El criterio de valoración compuesto específico renal se redujo en un 39% en el ensayo EMPA-REG, un 40% en el estudio CANVAS, un 47% en el estudio DECLARE-TIMI 58, un 34% en el ensayo CREDENCE y un 44% en el ensayo DAPA-CKD. La mayor sorpresa es el significativo efecto favorable de los SGLT2I en la mortalidad global de los pacientes con ERC con o sin DMT2. No se había encontrado con anterioridad un beneficio de supervivencia similar con ninguno de los medicamentos utilizados en pacientes con ERC diabéticos y no diabéticos. En esta revisión presentamos los resultados del ensayo DAPA-CKD, el ensayo CREDENCE y varios ensayos de resultados cardiovasculares (CVOT) que utilizaron diferentes SGLT2I y mostraron que los pacientes con niveles más bajos de tasa de filtración glomerular estimada (TFGe) pueden gozar de un mayor beneficio con respecto a la morbilidad cardiovascular que los pacientes con función renal normal. Además, se abordan los diferentes mecanismos de acción que explican los efectos renales beneficiosos de los SGLT2I.
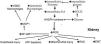
The glomerular ultra-filtrate delivered to the proximal convoluted tubules (PCT) within the kidneys has the same glucose concentration of plasma inflowing to the kidneys. The SGLT2 co-transporters located within the epithelial brush border of PCT S1and S2 segments reabsorb 90% of this glucose. Under normal conditions, SGLT2 together with SGLT1 within S3 PCT reabsorb all delivered glucose as long as its concentration is below 180mg/dL (renal threshold). With the onset of diabetes, increased blood sugar stimulates SGLT2 to increase renal threshold with consequent increase of sodium absorption by PCT. This leads to decreased delivery of sodium to the distal nephron segment to underlie the commonly observed glomerular hyperfiltration in diabetic patients (Fig. 1).1 It seems that increased renal threshold of glucose in T2DM is due to increased activity of existent transporters rather than an increase in the number of these co-transporters. In their distinguished work, Solini et al., have disclosed that the expression of SGLT2 and SGLT1 within the kidney tissue of T2DM patients is slightly lower compared with well-matched people without diabetes.2

Mechanism of hyperfiltration induced by hyperglycemia and how do SGLT2Is control it. UF: ultrafiltrate; SGLT: sodium glucose transporter; Na+: sodium; PCT: proximal convoluted tubules; DCT: distal convoluted tubules; ATP: adenosine triphosphate; MD: macula densa; AMP: adenosine monophosphate; VD: vasodilatation; AA: afferent arteriole; VC: vaso-constriction.
Phlorizin, a natural compound derived from apple parks was the first agent discovered to competitively inhibit both SGLT1 and SGLT2. By using this agent, urine glucose excretion increased in diabetic rats with instantaneous decrease of blood sugar. Inhibition of SGLT1 by phlorizin decreases intestinal glucose absorption and causes gastrointestinal upset associated with diarrhea. Moreover, bioavailability of phlorizin after oral intake is poor.3 During 2008, dapagliflozin (Dapa) was the first highly selective SGLT2I to be introduced4 followed by canagliflozin (Cana) and then empagliflozin (Empa).3 These agents reach their target on the brush border of PCT cells to similar extent via glomerular ultrafiltrate and tubular secretion.5
In 2012, Dapa was the first SGLT2I to get marketing authorization in Europe, followed 2 years later by FDA approval together with the other 2 SGLT2 inhibitors. In September 2015, the New England Journal of Medicine (NEJM) published the first large outcome trial, EMPA-REG. this trial disclosed the outstanding cardiovascular and renal benefits of Empa beside its hypoglycemic effect. The use of Empa was associated with a decrease of systolic blood pressure by around 4–5mm Hg, decrease of serum uric acid level by 0.6–0.8mg/dL, a 39% reduction in incident or worsening of diabetic nephropathy, a 38% reduction in the chance of progression to overt albuminuria (>300mg/day), a 44% reduction in the risk of doubling of serum creatinine, and a 55% reduction in the incidence of starting renal replacement therapy. This trial included 7020 patients in 50 sites. 25.5% of participants had CKD stage 3; 17.8% were in stage 3a and 7.7% were in stage 3b. 11% had overt albuminuria and approximately 80% were on a renin–angiotensin system (RAS) blocker. Beside the unexpected and marvelous renal outcome results, 3.1 years use of Empa achieved a 14% reduction of the composite of death from cardiovascular causes, non-fatal myocardial infarction or non-fatal stroke.6 EMPA-REG was the first to demonstrate a survival benefit in patients suffering DKD. In RENAAL trial, losartan succeeded to decrease the need for dialysis by 27% but there was no impact on patient survival.7 In similar trials, valsartan, irbesartan, candesartan and olmesartan failed to show survival benefit in diabetic nephropathy patients. On the other hand, Empa use was associated with 32% reduction in all cause mortality and 38% in cardiovascular mortality.6 Similarly, Cana use also decreased the risk of all cause mortality by 13% and cardiovascular mortality by 13%.8 The distinguished cardiovascular impact of SGLT2Is is a reflection of many therapeutic benefits unrelated to their hypoglycemic effect. The administration of Dapa for one year to 32 T2DM significantly decreased arterial stiffness assessed by carotid femoral arterial pulse wave velocity.9 Arterial pulse wave velocity is a better identifier of high risk population for cardiovascular disease.10 The reason behind the observable difference between EMPA-REG and CANVAS as regards the impact on mortality is likely due to the difference in the percentage of patients suffering established cardiovascular disease (CVD); while all patients in EMPA-REG trial had established CVD, only 66% of the cases studied in CANVAS had established CVD. This would explain the lack of significant effect of Dapa on overall or cardiovascular mortality in DECLARE-TIMI 58 trial that had only 59% of recruited cases with established CVD and 92.6% of the cases had basal estimated glomerular filtration rate (eGFR) more than 60mL/min/m2.11 Because EMPA-REG, CANVAS, and DECLARE-TIMI 58 trials were CVOT studies that were designed to examine the cardiovascular safety, the renal outcome parameters were secondary end points. Further studies were designed looking more thoroughly on the renal end points.
In the CREDENCE trial, T2DM patients having albuminuric CKD were assigned to receive Cana at a dose of 100mg daily or placebo. All the patients had an eGFR of 30 to <90ml/min/m2 and albuminuria >300–5000mg/g creatinine and were treated with RAS blockers. The primary outcome was a composite of end-stage renal disease (ESRD), doubling of serum creatinine, or death from renal or cardiovascular causes. The projected duration of the trial was 5.5 years, but it was prematurely terminated according to the recommendation of the data and safety monitoring committee after a median follow-up of 2.62 years thanks to the overwhelming efficacy of Cana. Patients in the Cana group had a highly significant reduction of the primary composite endpoint by 34%, a lower risk of ESRD, hospitalization for HF, and the composite of CV death, myocardial infarction, or stroke.12 In the last March 2020, DAPA-CKD trial was prematurely stopped following a recommendation from an independent Data Monitoring Committee (DMC) based on its determination of overwhelming efficacy of Dapa in CKD patients with and without diabetes. In this trial, 4304 participants with an eGFR of 25–75mL/min/m2 and a urinary albumin-to-creatinine ratio of 200–5000mg/g creatinine were randomly assigned to receive dapagliflozin (10mg once daily) or placebo. The primary outcome in this trial was a composite of a sustained decline in eGFR of at least 50%, ESRD, or death from renal or cardiovascular causes. Over a median of 2.4 years, the primary outcome event risk was reduced by 39%. The hazard ratio for the composite of a sustained decline in the eGFR of at least 50%, ESRD, or death from renal causes was reduced by 44%, the risk for the composite of death from cardiovascular causes or hospitalization for heart failure was reduced by 29%, and the overall mortality was reduced by 31% in Dapa arm. The more surprising in this trial is that the effects of Dapa were similar in participants with and without T2DM13 (Table 1). DAPA-CKD trial is the first trial in the history of Medicine that demonstrates survival benefit in CKD patients. Moreover, the results of CREDENCE and DAPA-CKD have confirmed that SGLT2Is can reduce cardiovascular and renal endpoints regardless of baseline eGFR. Patients with lower eGFR at exposure to SGLT2Is seem to have greater cardiovascular and renal protection than subjects with higher eGFR with respect to relative as well as absolute risk reduction.14 The action of Cana and Dapa in CKD patients took place in spite of the decreased availability of SGLT2 co-transporters within the chronically diseased kidneys. Nakamura et al., have demonstrated that the mRNA expression level of SGLT2 is markedly depressed in 5/6 nephrectomised compared to sham operated rats.15 In addition, a recent meta-analysis did show that patients with lower baseline eGFR have significantly lower risk for heart failure hospitalization than patients with higher baseline eGFR.16 Moreover, the number needed to treat for 5 years to prevent one renal event is 21 in patients with eGFR <45mL/min/1.73m2, while it is 30, 62 and 79 in patients with an eGFR 45–60, 60–90 and >90mL/min/1.73m2, respectively.14
Summary of the outcome of the five large-scale trials of SGLT2 inhibitors in T2DM, CKD, and heart failure.
| Trial name and design | Inclusion criteria | Main cardiovascular outcome | Main renal outcome |
|---|---|---|---|
| EMPA-REGNumber of patients 7020EMPA 10mg, EMPA 25mg, or placebo once dailyMean eGFR 74.1mL/min/1.73m2median follow-up: 3.1 years | Type 2 diabetesEstablished CVD (>99%)HbA1c 7.0–10.0%BMI ≤45kg/m2eGFR ≥30mL/min/1.73m2 | 14% reduction [HR 0.86; (95% CI 0.74–0.99), P 0.04] of composite of cardiovascular death, non-fatal myocardial infarction or non-fatal stroke | 39% reduction [HR 0.61; (95% CI 0.53–0.70), P<0.001] of renal-specific composite outcome |
| CANVASNumber of patients 10,142CANA 300mg, CANA 100mg or placebo once dailyMean eGFR 76.5mL/min/1.73m2Median follow-up: 2.4 years | Type 2 diabetesEstablished CVD (65.5%) or two or more risk factors for CVD (34.5%)HbA1c 7.0–10.5%eGFR ≥30mL/min/1.73m2 | 14% reduction [HR 0.86; (95% CI 0.75–0.97), P 0.02] of composite cardiovascular death, non-fatal myocardial infarction or non-fatal stroke | 40% reduction [HR 0.60; (95% CI 0.47–0.77)] of renal-specific composite outcome |
| DECLARE-TIMI 58Number of patients 17,160DAPA 10mg or placebo once dailyMean eGFR 85.2mL/min/1.73m2Median follow-up: 4.2 years | Type 2 diabetesEstablished CVD (40.5%) or two or more risk factors for CVD (59.5)HbA1c 7.0–10.5%eGFR ≥30mL/min/1.73m2 | 14% reduction [HR 0.86; (95% CI 0.75–0.97), P 0.02] of composite cardiovascular death, non-fatal myocardial infarction or non-fatal stroke | 40% reduction [HR 0.60; (95% CI 0.47–0.77)] of renal-specific composite outcome |
| CREDENCENumber of patients 4401CANA 100mg or placebo once dailyMean eGFR 85.2mL/min/1.73m2Median follow-up: 2.6 years | Type 2 diabetesEstablished CKD: eGFR 30–90mL/min/1.73m2 and UACR 200–5000mg/g≥30 years of ageHbA1c 6.5–12.0% | 31% reduction [HR 0.69; (95% CI 0.57–0.83), P<0.001] of composite of cardiovascular death or hospitalization for heart failure | 34% reduction [HR 0.66; (95% CI 0.53–0.81), P<0.001] of renal-specific composite outcome |
| DAPA-CKDNumber of patients 4304DAPA 10mg or placebo once dailyMean eGFR 43.2.2mL/min/1.73m2Median follow-up: 2.4 years | Adults with or without type 2 diabetesEstablished CKD: eGFR 25–75mL/min/1.73m2 and UACR 300–5000mg/gMean age 61.8±12.1 years | 29% reduction [HR 0.71; (95% CI 0.55–0.92), P<0.001] of composite of cardiovascular death or hospitalization for heart failure | 39% reduction [HR 0.61; (95% CI 0.51–0.72); P<0.001] of renal-specific composite outcome |
| DAPA-HFNumber of patients 4744DAPA 10mg or placebo once dailyNYHA class II, III, or IV with EF ≤40%median follow-up:18.2 months | Adults ≥18 years with or without type 2 diabetesHFrEF with EF ≤40%NT-proBNP ≥600pg/ml or history of hospitalization within past year+NT-proBNP ≥400pg/ml | 26% reduction [HR 0.74; (95% CI 0.65–0.85); P<0.001] of composite of worsening heart failure or cardiovascular death | No significant difference in the incidence of the prespecified renal composite outcome |
eGFR: estimated glomerular filtration rate; CVD: cardiovascular disease; BMI: body mass index; HR: hazard ratio; CI: confidence interval; renal-specific composite outcome: ≥40% decrease eGFR, new end-stage renal disease, or death from renal or cardiovascular causes; UACR: urine albumin/creatinine ratio; NYHA: New York Heart Association; NT-pro BNP: N-terminal pro b-type natriuretic peptide.
Accumulating evidence supports the opinion that the cardiac and renal protection offered by SGLT2Is is not related to their hypoglycemic effect. The lower risk of the primary outcome overall as well as ESRD in patients receiving Cana than those taking placebo in CREDENCE trial occurred despite very modest between-group differences in blood glucose level.12 Further three studies have shown beneficial effects of SGLT2Is in spite of attenuated effects on HbA1c.17–19 Systemic effects of SGLT2Is help intra-renal mechanisms of renal protection. Systemic effects include reduction of body weight, systemic blood pressure and systemic inflammation.20
Control of glomerular hyperfiltrationThe first and most acceptable intrarenal mechanism is the control of glomerular hyperfiltration by SGLT2Is. Inhibition of sodium and glucose uptake by PCT increases sodium delivery to the distal segment. The macula densa within the distal convoluted tubules (DCT) interprets increased sodium delivery as circulating volume expansion and through tubuloglomerular feedback cause hemodynamic changes in the afferent arterioles. Administration of Empa to diabetic rats cause vasoconstriction of afferent arteriole within 30min together with decreased intraglomerular tuft pressure and reduction of single nephron GFR. Adenosine A1 receptors blocking abolished these effects.21 Adenosine increases as a consequence of excess ATP consumption during increased active sodium reabsorption by DCT (Fig. 1). The impact of SGLT2Is on GFR can occur even in euglycemic state. SGLT2Is reduced the renal glucose concentration threshold similarly in T2DM and non-diabetic individuals to less than 40mg/dL.22 Increased single nephron ultrafiltration of the surviving nephrons is one of the main causes of progressive decline in kidney function in CKD patients. By controlling single nephron hyperfiltration, SGLT2Is can mitigate CKD progression even in non-diabetic patients. In view of the observed decrease of glomerular hyperfiltration after use of Dapa in T2DM patients in spite of the unchanged renal vascular resistance, a recent study suggested that the decrease in glomerular tuft pressure is more likely caused by post-glomerular efferent arteriolar vasodilatation instead of preglomerular afferent arteriolar vasoconstriction. The authors hypothesized that the potential vasoconstrictive action of adenosine on the afferent arteriole was counteracted by a concomitant RAS blockade or an increase in prostaglandin synthesis. Subsequently, prostaglandins may have induced efferent arteriolar vasodilation, which could not be prevented by angiotensin II, since this was pharmacologically blocked.23
Uric acid wastingThe kidney eliminates 70% of the daily UA production in humans.24 The offending effect of high serum uric acid (SUA) on the kidney was clearly demonstrated in experimental animals after raising their SUA. Animals have low SUA due to the uricase enzyme that breaks down UA. Oxonic acid inhibits the uricase enzyme and is used in experimental animals to raise their SUA. By increasing SUA, experimental animals develop systemic hypertension, glomerular hypertension, glomerulosclerosis, and interstitial fibrosis.25–29 SUA is a strong predictor of increased urine albumin excretion. On follow-up of T1DM patients having normal urine albumin for 6 years, every 1mg/dL SUA above normal level increases the risk of development of albuminuria by 80%.30 The risk for albuminuria and accelerated decline of GFR in T2DM patients suffering hyperuricemia is further illustrated by 2 prospective studies.31,32 In patients suffering T2DM for fifteen years or more, SUA >7mg/dL in males and >6mg/dL in females increases the risk of DKD progression, and overall mortality.33 Treatment of patients suffering T2DM and DKD and high SUA with allopurinol decreased albuminuria and serum creatinine significantly over three years of follow-up.34 A prospective observation study of 900 blood donors followed for 5 years showed that the basal SUA is a significant predictor of eGFR decline in healthy normotensive subjects lacking signs of CKD at entry to the study.35 In contrast, recent studies failed to support the therapeutic benefit of urate lowering treatment. Allopurinol failed to improve kidney outcomes among patients with T1DM and early-to-moderate DKD.36 Treatment of hyperuricemia using allopurinol also failed to slow the rate of decline in eGFR as compared with placebo in patients with CKD and a high risk of progression.37 Similarly, febuxostat failed to mitigate the progression of CKD among patients with asymptomatic hyperuricemia and stage 3 CKD.38 Based on clinical and experimental evidence, it seems that intracellular UA behaves differently in comparison to extracellular UA. While extracellular UA has anti-oxidant properties, intracellular UA imposes a detrimental pro-oxidant effect.39
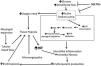
Increased intracellular production of uric acid (UA) occurs in epithelial cells of PCT in diabetic state. Excess glucose absorption by PCT in diabetic patients increases intracellular glucose availability. A consequent increase in the activity of polyol pathway leads to increased fructose synthesis. Fructose metabolism leads to increased intracellular adenosine and UA synthesis.40 Increased intracellular UA stimulates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme causing increased intracellular oxidative stress, mitochondrial injury, adenosine triphosphate (ATP) depletion,41,42 endothelial injury, RAS activation and increased epithelial- mesenchyme transition (EMT). Increased EMT leads to overproduction of fibroblasts that infiltrate the interstitium with consequent progressive interstitial fibrosis.43 Increased NADPH oxidase activity can also stimulate production of macrophage chemo-attractant factor (MCP1) with consequent increased macrophage infiltration of the kidney44 (Fig. 2). By inhibiting glucose absorption, SGLT2Is decrease intracellular glucose concentration, decrease activity of polyol pathway, decrease fructose and thus decrease intracellular UA synthesis.45 Moreover increased availability of luminal glucose as a consequence of SGLT2Is administration triggers apical GLUT9 isoforms in PCT epithelium trying to absorb glucose in exchange with UA. GLUT9 activation thus depletes intracellular UA within PCT. In contrast, GLUT9 in the collecting ducts increase absorption of uric acid from the lumen. Excess glucose within the tubular lumen inhibits UA absorption by GLUT9 in the collecting ducts46 (Fig. 3). Contrary to the debatable impact of serum UA reduction, it seems that reduction of intracellular UA within PCT have a crucial role in the renal protection of SGLT2Is. Among the different angiotensin receptor blockers, only losartan has the ability to lower serum UA through its uricosuric effect.47 Decreased uptake of UA by PCT cells result in decreased intracellular UA within these cells. Losartan is the only member among its family that showed a reduction for the need for dialysis in patients suffering DKD.7

Different pathogenic mechanisms of kidney injury possibly induced by uric acid. UA: uric acid; ROS: reactive oxygen species; NF-κB: nuclear factor kappa B; MCP1: macrophage chemo-attractant protein-1; RAS: renin angiotensin system; EMT: epithelium mesenchyme transition; VSMC: vascular smooth muscle cells.

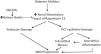
Sodium hydrogen exchangers (NHE) exist in nine isoforms.48,49 NHE3 isoform is encountered on renal tubular and intestinal epithelium. When NHE3 in PCT and ascending loop of Henle are activated, excess sodium retention occurs and contributes to systemic hypertension in diabetic patients.50,51 Diabetes and excess insulin stimulate while SGLT2Is inhibit renal NHE351 (Fig. 4). The natriuretic effect of SGLT2Is is mediated mainly through NHE3. Empa failed to induce natriuresis in normo-glycemic NHE3 knock out mice in contrast to wild mice.52 The natriuretic effect of SGLT2Is is responsible for the drop of blood pressure observed after the use of these agents. In salt treated obese and metabolic syndrome rats that develop hypertension, treatment with SGLT2Is significantly decreased the blood pressure associated with an increase in urinary sodium excretion. This was associated with normalized circadian rhythms of both blood pressure and sympathetic nerve activity and might explain why the observed hemoconcentration induced by SGLT2Is is not associated with change in heart rate.53

Increased activity of NHE3 isomer within the proximal convoluted tubules increases sodium absorption from the lumen of these tubules in exchange with the secreted hydrogen. Decreased sodium delivery to the distal nephron segments results in glomerular hyperfiltration. Diabetic state and insulin administration increase NHE3 activity while SGLT2Is and GLP1RAs inhibit it. NHE: sodium hydrogen exchanger; SGLT2Is: sodium glucose transporter-2 inhibitors; GLP1Ras: glucagon like peptide receptor agonists.
NHE1 isoform present on the surface of cardiomyocytes, endothelial cells, and platelets are also stimulated in diabetic patient and are inhibited by SGLT2Is. The favorable impact of SGLT2Is on the heart and endothelium are mediated, in part, through NHE1 inhibition.50,51
Anti-inflammatory effects of SGLT2IsPCT epithelial cells incubated in high glucose medium increase the expression of Toll-like receptor-4, increase nuclear DNA binding for NF-κB and activator protein 1, and increase collagen IV expression as well as interleukin-6 secretion. All these processes are inhibited on addition of Empa to the culture medium.54 SGLT2Is inhibit inflammation indirectly through suppression of oxidative injury. In diabetic kidney, generation of free radicals is a result of a pro-oxidant enzyme action, polyol pathways, lipid peroxidation, mitochondrial dysfunction, hemodynamic changes, and protein kinase C activation. Empa inhibits free oxygen radical production through inhibition of NADPH oxidase enzyme.55 Other pro-oxidant enzymes like endothelial nitric oxide synthase, and xanthine oxidase, were also modified by different SGLT2Is through alteration of their activity or their expression.56,57 Suppression of free oxygen radicals consequently suppresses MCP1 and NFκB. High mobility group box protein 1 (HMGB1) is a nuclear DNA-binding protein normally indulged in a variety of processes including repair, differentiation, and development of DNA. Under pathological circumstances, HMGB1 is released from the necrotic or activated cells and acts as a proinflammatory cytokine. HMGB1 exerts its functions via several receptors including RAGE and toll-like receptors (TLRs).58 Macrophages and activated dendritic cells transport HMGB1 from the nucleus to the cytoplasm and then release it via secretory inflammasomes.59 Through the interaction with TLR2/4 receptors, HMGB1 augments the translocation of NF-κB p65 subunit into the nucleus to promote synthesis of different inflammatory mediators.60 HMGB1 plays a crucial role in the pathogenesis of inflammation in diabetic patients.61 When Empa was administered for 4 weeks to male Wistar rats 8 weeks after induction of diabetes, renal cortical NF-κB was down regulated together with reduction of renal levels of HMGB1, RAGE, and TLR-4.62
Promotion of autophagyAutophagy is an intracellular lysosome-dependent process that sweeps dysfunctional organelles out of the cytoplasm. Increased accumulation of dysfunctional organelles is a common feature of diabetic kidney as a sequence of increased oxidative stress and endoplasmic reticulum stress. These accumulating organelles pose changes in ion channels and trigger cellular inflammation and thus endanger cell survival. Autophagy is usually mitigated in diabetic patients and in cases receiving insulin. Impaired autophagy is due to deficiency of sirtuin-1 (SIRT1), and AMP-activated protein kinase (AMPK) in diabetic patients and manifests as decrease of autophagy of the podocytes and the renal tubular cells.63 By mimicking fasting state, SGLT2Is augment AMPK/SIRT1 signaling and stimulate autophagy, thereby decrease the accelerated damage of podocytes and renal tubular cells within the kidney.64
Reversal of renal hypoxiaChronic renal hypoxia is recently suggested as an important cause of renal damage encountered in DKD patients. This hypoxia is the consequence of a mismatch between oxygen delivery and oxygen demand. Decreased oxygen delivery is the result of microangiopathy affecting small intra-renal arteries together with the glomerular mesangial expansion that compromises the down stream tubular blood flow. Increased oxygen demand evolves as the result of increased activity of the sodium pumps in parallel with sodium glucose cotransporters up regulation and to face glomerular hyperfiltration. Once the imbalance between demand and delivery exists, subsequent tissue hypoxia causes capillary damage, extracellular matrix expansion, inflammation, tubular damage, and subsequent fibrosis. Nephron loss increases the workload of surviving units. This exaggerate the delivery/demand of the surviving nephrons and thus ensuing a vicious cycle that exaggerates subsequent damage.65 Tissue hypoxia stimulates hypoxia inducible factor (HIF) expression in renal tissue. HIF plays an important role in hypoxia-induced tubulointerstitial fibrosis. SGLT2Is inhibit HIF gene expression, and thus inhibit hypoxia induced renal fibrosis.66 This effect of SGLT2Is is likely related to improved tissue hypoxia thanks to the significant down regulation of oxygen consumption by PCT cells after SGLT2Is administration. Further renoprotection offered by SGLT2Is is related to their anti-inflammatory effect of these agents discussed above.54 As we discussed above, free oxygen radical injury causes mitochondrial damage and depletion. A recent study has demonstrated that the SGLT2I Dapa improves mitochondrial function. Dapa increases a panel of urinary metabolites linked to mitochondrial metabolism.67 Decreased sodium and glucose uptake by the PCT can thus reduce cortical oxidative stress and enhance recovery from tubulointerstitial damage that possibly restores renal erythropoietin (EPO) production. Increased erythropoiesis significantly improves hematocrit that improves tissue oxygen delivery68 (Fig. 5).
Enhanced ketogenesis
Decreased pancreatic insulin secretion and/or decreased insulin requirement after SGLT2Is administration render circulating insulin not enough to suppress lipolysis and ketogenesis. Increased pancreatic secretion of glucagon induced by SGLT2Is further increases ketogenesis.69 SGLT2Is mediated cardiac protection is related, in part, to enhanced ketogenesis.70 ATP production within epithelial cells of PCT is largely dependent on fatty acid oxidation that is suppressed in diabetic patients. Impaired ATP production is responsible for progression of CKD and DKD.71 Ketone bodies are an energy source substitute that is capable to ameliorate defective energy metabolism in the epithelial cells of PCT and decrease the progressive loss of PCT and fibrosis after administration of SGLT2Is. In addition to acting as direct energy source for damaged PCT instead of deficient fatty acids, ketone bodies prevent PCT epithelial and podocyte damage by inhibiting the mammalian target of rapamycin complex 1 (mTORC1) signaling. The last mechanism might explain the renoprotective value of SGLT2Is in normoproteinuric as well as proteinuric CKD72 (Fig. 6).

Ketone bodies as inhibitors of the renal mammalian target of rapamycin C1. Through this mechanism, SGLT2 inhibitors protect the kidney against the offending action of mammalian target of rapamycin that is induced by diabetic state. SGLT2Is: sodium glucose co-transporter inhibitors; PCT: proximal convoluted tubules; DKD: diabetic kidney disease.
Up-regulation of SGLT2 by hyperglycemia results in accumulation of glucose within the cytoplasm of PCT cells with consequent aberrant glycolysis. Accumulating glycolytic products are thus diverted into four noxious pathways, namely, the polyol pathway to produce sorbitol and fructose, the hexosamine pathway to form hexosamine that is responsible for formation of transcription factor SP1 and transforming growth factor-β (TGF-β1) induction,73 the lipid synthesis pathway to form diacyl glycerol that induces protein kinase c (PKC) activity, and the formation of advanced glycation end-products. These paths eventually lead to overproduction of TGF-β1 and IL-8, as well as epithelial mesenchymal transition (EMT) of PCT epithelial cells and endothelial mesenchymal transition (EndMT) in the adjacent capillaries. These events eventually lead to interstitial inflammation and fibrosis. By inhibiting excess cellular glucose uptake, SGLT2Is abort all this cascade of events as proved by in- vitro and experimental animal studies.74,75
Prevention of renal senescencep21Cip1/Waf1 is a cyclin-dependent kinase inhibitor that increases in epithelial cells of PCT in patients suffering diabetes. This effect is aborted by knocking down SGLT2.76,77 These findings suggest that hyperglycemia causes PCT cell senescence via a SGLT2- and p21-dependent pathway in the diabetic kidney. SGLT2Is reduce the increased expression P21 and thus decrease senescence of renal tubular cells76,77 (Fig. 7).

Activation of SGLT2 in diabetic patients leads to overactivity of P21, the natural inhibitor of cyclin-dependent kinase 2. This kinase enzyme inhibits cell senescence. By inducing P21, diabetic patients suffer increased proximal tubular epithelium senescence. Through inhibition of SGLT2, SGLT2Is protect proximal tubular epithelial cells against increased senescence. SGLT: sodium glucose transporter; PCT: proximal convoluted tubule.
Despite the widespread use of different therapies for DKD and non- diabetic CKD, including statins and RAS blockers, over the past decades, there is still a substantial residual burden of CVD. The significant reduction in cardiovascular adverse events and progression to ESRD associated with the use of SGLT2Is is a distinguished advantage of this novel group of hypoglycemic agents. The most outstanding cardiovascular effect of this group is the highly significant reduction in the incidence and hospital admission for heart failure among the wide spectrum of urine protein excretion and GFR. The cardioprotective benefits of SGLT2Is are multifactorial, including blood sugar control, body weight reduction, systolic blood pressure reduction, diuretic effect, hypouricemic effect, improved erythropoiesis, reduced sympathetic nerve activity,53 and inhibition of sodium hydrogen exchangers. Microalbuminuria was found associated with the future development of heart failure with reduced ejection fraction (HFrEF) and with systolic dysfunction in a community-based sample. On the other hand, CKD was modestly associated with heart failure whether having HFrEF or heart failure with preserved ejection fraction (HFpEF).78 Conversely, heart failure increases the risk of renal function decline and of adverse renal outcomes.79
SGLT2Is have also a significant favorable effect on arterial stiffness.9 Increased pulse wave velocity, as an index of arterial wall stiffness, is associated with a higher risk of incident CKD. The greater the carotid artery stiffness, the higher is the chance of incident CKD and the steeper is the rate of annual GFR decline.80
These cardiovascular benefits offered by SGLT2Is in diabetic and non-diabetic CKD patients might cast their impact on the improved renal outcome in these patients.
Anti-proteinuric effectProteinuria is recognized as a predictor of the decline in glomerular filtration rate. The higher the protein excretion rate the more likely the expected severity of renal disease as well as its rate of progression. Independent of the initial insult, different causes of CKD common pathogenic mechanisms that lead to single nephron hyperfiltration, proteinuria, progressive tubular injury, and renal scarring. Protein leakage into the glomerular ultrafiltrate stimulates excess proximal tubular protein reabsorption. Excess protein load within PCT epithelial cells triggers tubular chemokine expression and complement activation. As a consequence, the interstitium of the kidney become a site for inflammatory cell infiltration and subsequent fibrosis. For this reason, proteinuria is considered a valuable surrogate end point for clinical trials in CKD patients and a target for renal protection studies.81 To look for the anti-proteinuric effect of Dapa, a multicenter randomized, double-blind, placebo-controlled crossover trial was done in adult non-diabetic CKD patients having 24h protein excretion between 0.5 and 3.5g/day and eGFR ≥25ml/min/m2 (DIAMOND study). In this trial, six weeks treatment of 52 patients with Dapa 10mg daily failed to achieve a significant decrease in urine protein excretion.82 The short duration of treatment might underlie this failure. In DAPA-CKD study, administration of 10mg Dapa for a mean period of 2.4 years was associated with a highly significant decline of urine protein excretion rate.13 The anti-proteinuric effect of Dapa was also demonstrated in a pooled analysis of 11 phase 3 randomized controlled clinical trials. These studies included 136 diabetic patients having urine albumin/creatinine ratio (UACR) ≥30mg/g that received 5 or 10mg Dapa in comparison to 69 patients that were kept on placebo. eGFR of these cases ranged between 11 and 45ml/min/1.73m2. Over 102 weeks, Dapa 5mg and Dapa 10mg reduced UACR by 47% and 35% respectively compared to placebo. There was no appreciable difference in glycemic control between the Dapa groups and the control cases.83 This is the first report of renoprotective effect of SGLT2Is in CKD patients having eGFR below 25ml/min/1.73m2.
A novel animal study has demonstrated that SGLT2 is expressed in mice podocytes. Induction of proteinuria in these mice was associated with upregulation of SGLT2 in podocytes. Dapa provided glomerular protection in these mice and limited proteinuria, podocyte dysfunction and loss induced by SGLT2 upregulation.84
Adverse events and risk/benefit profile of SGLT2IsThe most common adverse event attributable to SGLT2Is is the increased risk of fungal and to a lesser extent bacterial genital infection. This complication significantly decreases with the optimum health care of the external genitalia.85 The chance to develop diabetic ketoacidosis (DKA) is higher in T1DM receiving SGLT2Is. This event is infrequent in T2DM, and this chance is still uncommon in cases receiving SGLT2Is. DKA complicating SGLT2Is is usually euglycemic.86 The risk of amputation observed in CANVAS trial was not observed in CREDENCE trial or in trials using other SGLT2Is. However, attention to lower limb ischemia should be done before administration and on regular basis in patients using SGLT2Is.
Factors that can modify the renal expression of SGLT2As discussed above, hyperglycemia increases SGLT2 activity in spite of the decreased expression. On the other hand, chronic kidney disease decreases SGLT2 protein expression. Experimental studies have shown that pharmacological SGLT2 inhibition in mice increases renal SGLT2 protein expression in spite of the inhibition of the physiologic uptake of glucose.87 The mechanisms of SGLT2 upregulation by SGLT2Is and in response to diabetes remain poorly understood. Studies in human embryonic kidney cells indicated that insulin stimulates SGLT2 activity.88 In cultured human proximal tubular cells, insulin, but not high glucose, enhances SGLT2 protein expression. This was associated with an increased reactive oxygen species within PCT epithelial cells.89 While hyperinsulinemia may contribute to the increased SGLT2 protein expression and activity in T2DM, it cannot explain the increased renal SGLT2 expression in hypoinsulinemic T1DM.87 Other molecules known to up regulate SGLT2 activity include protein kinase C and protein kinase A, while angiotensin II AT1 receptors and hepatocyte nuclear factor-1α increase SGLT2 expression in the diabetic state.90
PerspectivesAlthough SGTL2Is received initial approval for glycemic control by the FDA in 2013, these agents proved, over a very short time, as effective agents for optimization of cardiovascular and renal outcomes in T2DM patients, and in non-diabetic patients. In addition to their striking benefits on the cardiovascular and renal outcome, their favorable impact on survival of these patients should reinforce the appealing attitude toward the usage of these agents for prevention of renal and cardiovascular complications even in non-diabetic CKD patients. Beside their use in prevention of cardiovascular and renal complications in T2DM, non diabetic patients with proteinuria, impaired kidney function, or CVD with HFrEF or HFpEF will also benefit on using these agents. The assessment of CVD and CKD risk status in every T2DM since their first presentation became a mandate. Estimation of GFR and urine albumin/creatinine ratio should be encouraged in various care settings, in addition to the use of validated risk scores for the estimation of atherosclerotic cardiovascular disease, heart failure, and CKD progression.91 The burden of polypharmacy on prescription is of significant concern in patients with T2DM, CKD, and CVD. An example is the use of SGLT2Is in patients using diuretics. Future may witness more ambitious clinical trials looking for the impact of the combination of SGLT2Is with other agents that showed promising results in recent trials like the nonsteroidal, selective mineralocorticoid receptor antagonist, finerenone, and/or the selective endothelin A receptor antagonist, atrasentan.
Finally we have to affirm that there is a precious opportunity to meaningfully reduce morbidity, mortality, and healthcare expenditures for different vulnerable patient groups including T2DM, CKD, and heart failure population.
KeypointsIn this review, we highlighted the different mega trials that disclosed the outstanding impact of the three famous sodium-glucose co-transporters 2 inhibitors (SGLT2Is) on chronic kidney disease (CKD) progression in diabetic and non-diabetic patients. We will try, in addition, to illustrate the different mechanisms that add to the beneficial effects of these agents in prevention and or withholding the progression of CKD.
Authors’ contributionsUsama Sharaf El Din and Mona Mansour Salem performed the majority of writing the manuscript.
Dina Ossama Abdulazim collected the references and highlighted the points of relevance in each article and revised the manuscript after being prepared.
FundingThis work did not receive funds or grants.
Conflict of interestAuthors declare no conflict of interests for this article.



