



Incidence of acute kidney injury (AKI) remained relatively stable over the last decade and the adjusted risks for it and mortality are similar across different continents and regions. Also, the mortality of septic-AKI can reach 70% in critically-ill patients. These sole facts can give rise to a question: is there something we do not understand yet?
Currently, there are no specific therapies for septic AKI and the treatment aims only to maintain the mean arterial pressure over 65mmHg by ensuring a good fluid resuscitation and by using vasopressors, along with antibiotics. On the other hand, there is an increased concern about the different hemodynamic changes in septic AKI versus other forms and the link between the gut microbiome and the severity of septic AKI. Fortunately, progress has been made in the form of administration of pre- and probiotics, short chain fatty acids (SCFA), especially acetate, and also broad-spectrum antibiotics or selective decontaminants of the digestive tract in a successful attempt to modulate the microbial flora and to decrease both the severity of AKI and mortality.
In conclusion, septic-AKI is a severe form of kidney injury, with particular hemodynamic changes and with a strong link between the kidney and the gut microbiome. By modulating the immune response we could not only treat but also prevent severe forms. The most difficult part is to categorize patients and to better understand the key mechanisms of inflammation and cellular adaptation to the injury, as these mechanisms can serve in the future as target therapies.
La incidencia de la lesión renal aguda (LRA) se ha mantenido relativamente estable a lo largo de la última década, con unos riesgos ajustados de padecer y morir a consecuencia de esta enfermedad similares en los distintos continentes y regiones. La mortalidad asociada a la LRA secundaria a sepsis puede llegar a 70% en los pacientes que se encuentran en estado crítico. Estos hechos, por sí mismos, deben llevarnos a plantearnos la siguiente pregunta: ¿se nos escapa algo que aún no comprendemos?
Actualmente no se dispone de terapias específicas para la LRA secundaria a sepsis y el tratamiento se centra únicamente en mantener la presión arterial media por encima de los 65mmHg mediante una rehidratación adecuada, vasopresores y antibióticos. Asimismo, cada vez existe mayor interés por las diferentes alteraciones hemodinámicas que se producen en comparación con otras formas de la enfermedad, así como por la relación existente entre el microbioma intestinal y la gravedad. Afortunadamente, se ha avanzado notablemente en la forma en la que se administran los prebióticos y los probióticos, los ácidos grasos de cadena corta (AGCC), especialmente el acetato, los antibióticos de amplio espectro o los detoxificantes selectivos del tracto digestivo, en un intento exitoso de modular la flora microbiana y disminuir tanto la gravedad de la LRA como su mortalidad.
En conclusión, la LRA secundaria a sepsis es una forma grave de lesión renal que provoca unos cambios hemodinámicos específicos y en la que se observa una estrecha relación entre la función renal y el microbioma intestinal. La modulación de la respuesta inmunitaria no solo permitiría tratar esta enfermedad, sino también prevenir las formas graves de la misma. La parte más difícil de este enfoque radica en clasificar correctamente a los pacientes y comprender mejor los mecanismos clave de la inflamación y la adaptación celular a la lesión, ya que estos pueden convertirse en futuras dianas terapéuticas.
In 1909, Sir William Osler used for the first time the expression “acute Bright's disease”, describing an acute form of nephritis and considering that the causes were trauma, toxic agents, excessive physical exertion and pregnancy.1 Since then, more and more descriptions that seemed to point to the same affection – a poor kidney function, have appeared, culminating in 2006 with the emergence of the term “acute kidney injury”(AKI) in order to “include epidemiological data and present it as a public health problem”.2
AKI refers to a rapid decrease in kidney function leading to altered homeostasis (retention of nitrogen waste products and acid–base and electrolyte disturbances). It is defined by Kidney Disease Improving Global Outcome (KDIGO) guidelines either as an increase in serum creatinine by a minimum of 0.3mg/dl in 48h (or to a minimum of 1.5 times baseline which is known or presumed to have occurred in the previous 7 days) or as a urinary volume under 0.5ml/kg/h for 6h.3 Also, there are three severity stages, the last often requiring kidney replacement therapy (KRT). Despite a few limitations of this definition – the lack of inclusion of the etiology of AKI, which is of the utmost importance in the management, the use of creatinine, which can be lower in septic AKI (without a major change in body-mass index, hematocrit or extracellular fluid), and therefore over-estimating the kidney function, and the urinary volume, which can be low only because of insufficient fluid resuscitation, it provides a generally accepted definition which can permit a rapid and easy diagnosis and management.3,4
Sepsis is defined by the Society of Critical Care Medicine/European Society of Intensive Care Medicine (SCCM/ESICM) task force as a “life-threatening organ dysfunction caused by a dysregulated host response to infection”, where organ dysfunction means a minimum of 2 points in the SOFA score (Sepsis-related Organ Failure Assessment Score) and infection is clinically and/or microbiologically demonstrated.5 Septic shock is a more severe stage of sepsis when the patient does not respond hemodynamically to fluid resuscitation and there is the need to add vasopressors in the treatment to maintain the mean arterial pressure above 65mmHg, the central venous pressure between 8 and 12mmHg and lactate under 2mmol.3,5
A few epidemiological observations: a number sometimes can tell more than a thousand wordsThere are a few facts of utmost importance. First of all, “incidence of AKI in the general population remained relatively stable over the last decade”,6 secondly, the incidence of AKI can reach 67% in critically-ill patients from intensive care unit (ICU) wards, septic AKI being the main cause of this category, with in-hospital mortality ranging from 21% to 70%,7,8 and last, but not least, a study conducted in Mayo Clinic Hospital over 9 years argued that “adjusted risks for AKI and mortality were similar across different continents and regions”.9
These arguments prove that almost nothing has improved in the last decade in the management of critically-ill patients from the point of view of preventing or treating AKI in general, and septic AKI in particular. Moreover, because the risk of mortality is similar worldwide, in developing countries as in more developed ones, it gives rise to a question: is there something about the pathophysiological mechanisms of septic AKI that we do not know yet?
Risk factors and prognosis of AKI or how a simple “injury” can transform into a lifetime disease (and not only one)There are some well-known risk factors for AKI that should be kept in mind, especially in critically-ill patients, in an attempt to prevent the development of kidney injury: sepsis, coronary artery disease, chronic liver disease, chronic kidney disease (CKD), nephrotoxic medication and the use of vasopressors.10 Regarding the prognosis, AKI should not be perceived as a temporary and limited event, but on the contrary, as the first step to a multitude of health problems: increased cardiovascular morbidity and mortality, new-onset CKD, progression of pre-existent CKD and an increased risk of end-stage kidney disease (ESKD).11–13 In addition, Venkatachalam and Murugan claimed that AKI increases 13 times the risk for CKD and CKD increases 7 up to 10 times the risk for AKI, and therefore the relationship between AKI and CKD is a bidirectional one.7,14
The proposed mechanisms through which AKI induces all these changes are vascular injury, glomerular hyperfiltration and interstitial fibrosis. The consequence is an irreversible loss of nephrons and, thus, a lower kidney survival, with all the burden and complications involved.15 As expected, the most affected will be the elderly due to low nephron reserve and comorbidities.16 However, even if AKI seemed to have passed without immediate consequences, it is advisable to perform at least once time per year a check for albuminuria, as a marker for CKD, even if the glomerular filtration rate (GFR) recovered completely17 and maybe a renal ultrasound too, as a less well-known long-term consequence of AKI is renal cancer through DNA damage and clonal expansion of mutated cells during the recovery phase.16 However, there are no clear epidemiological data in the literature about this last possible complication.
Sepsis-induced AKI: mechanisms that stand behindThere are a lot of theories behind septic AKI, many of them focusing on three parts: hemodynamic factors leading to microvascular dysfunction, inflammation and cellular and metabolical responses to the injury that will be revised here.
First of all, as early as 1977, Ravikant and Lucas found on a pig model that renal blood flow (RBF) is not decreased, but on the contrary, even increased, both globally and in the kidney medulla during hyperdynamic sepsis.18 However, it was not an observation made on a human model, and therefore a few years passed until in 1990 Brenner et al. demonstrated that RBF is normal in septic AKI using eight critically-ill patients with an indwelling thermodilution renal vein catheter, but sepsis-induced kidney dysfunction occurs.19 Despite the small number of patients, these findings were consistent with observations made on later animal models, in 2006 Langenberg et al. arriving at the same conclusions using another animal model, this time seven Merino sheep.20 In 2008, Wan et al. concluded, based on all these observations that maybe it was the time to shift the paradigm in septic AKI from vasoconstriction and ischemia to vasodilatation with hyperemia, septic AKI being a type of AKI with renal vasodilatation, increased RBF, but also with increased serum creatinine and decreased GFR.21
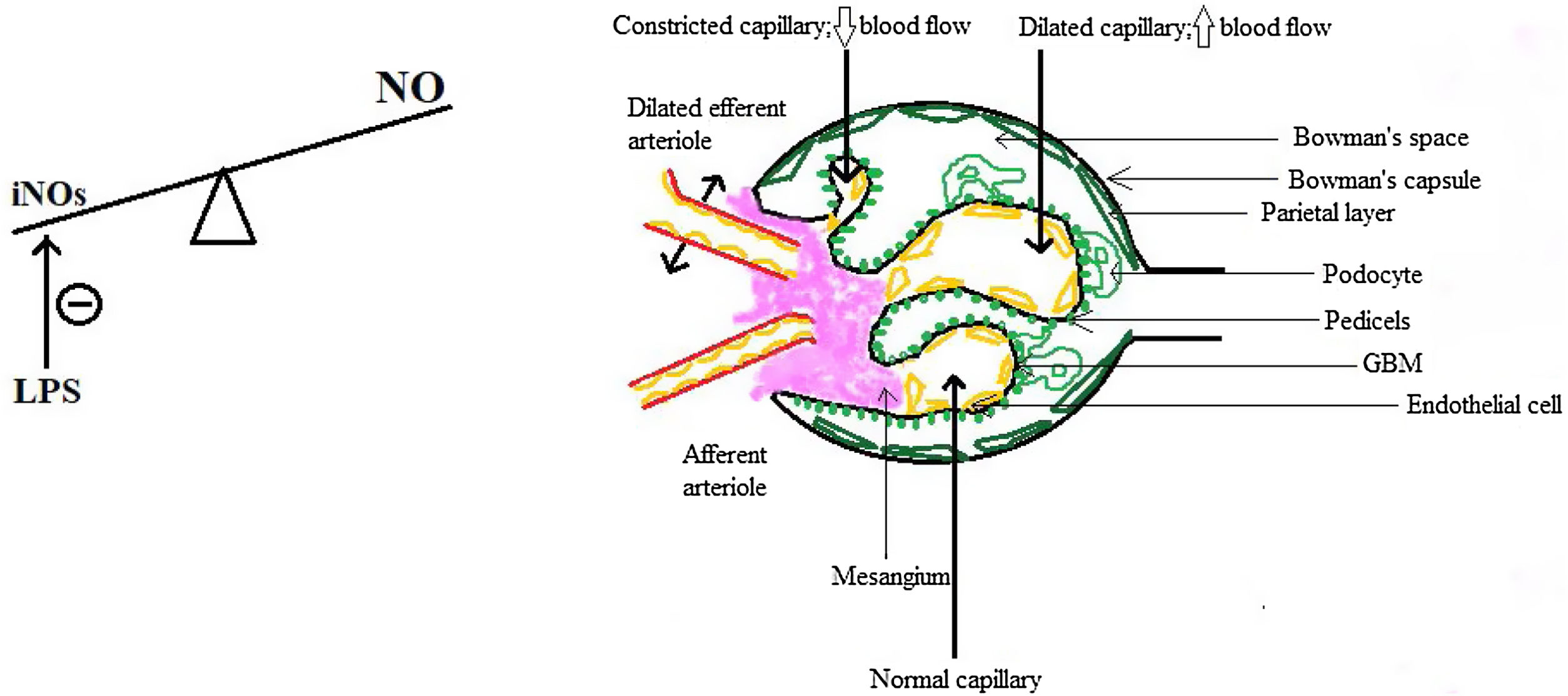
So the question arises: if the kidneys are not just well-perfused, are even over-perfused, why does the GFR drops? The answer resides, but only in part, in the intra-renal hemodynamical changes. Gomez and Kellum made in 2016 a series of interesting observations about renal microcirculation during septic AKI, claiming that there are ischemic areas alternating with hyperemic ones, a fact that could explain why the GFR drops and RBF is maintained.22 The key role in this concept is played by the nitric oxide (NO) and by the inducible NO synthase (iNOs), because in sepsis there is an increase in total NO but a heterogenous distribution of iNOs which is also locally inhibited by the lipopolysaccharide (LPS),23 therefore leading to an increase in the number of capillaries with blood flow deficit.22,24 Moreover, there is a difference between cortical and medullary renal flow: cortical blood flow was measured by laser Doppler and found to be decreased and also the medullary flow did not redistribute to kidney cortical, despite the need.23 The GFR drops also because of the predominant efferent arteriole vasodilatation, which causes a decrease in hydrostatic pressure in glomerular capillaries, some authors regarding this as a protective mechanism since a lower filtration rate means less exposure to toxins such as damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) and less energy consumption for the reabsorption of substances.25 Finally, there are several changes in the capillary structure: endothelial dysfunction, with barrier alteration and enhancement of vascular permeability, in part due to pericyte detachment,26,27 interstitial edema and glycocalyx impairment, all of which predispose to sluggish intraglomerular and peritubular blood flow, intraglomerular thrombosis and prolongation of the time inflammatory mediators interact with renal cells,21,22 which opens the gate for the participation of the inflammation in the pathogenesis of septic AKI.
These aspects are depicted in Fig. 1.



Inflammation is a constant component of each microbial injury leading, ideal, to overcoming the insult through a perfect equilibrium between pro-inflammatory status and the anti-inflammatory one.28 However, not a few times the host has these mechanisms disrupted and therefore bacteremia and sepsis can occur. The pro-inflammatory status comprises, broadly, of activation of the complement and the coagulation, activation of proteases, release of reactive oxygen species and cytokines like IL-1, IL-6, platelet-activating factor (PAF), tumor necrosis factor alpha (TNF-α) and last, but not least, cellular involvement – neutrophils, monocyte/macrophages, thrombocytes and endothelial cells.28 The anti-inflammatory status implies increased levels of IL-10, inhibition of phagocytosis, impairment of chemotaxis and apoptosis of the lymphocytes.28 The consequences are of great severity and can lead to the death of the host: mitochondrial and endothelial dysfunction, apoptosis and necrosis, capillary leak, thrombosis, oliguria, AKI, multi-system organ failure (MSOF) and, in the end, death.28 On the other hand, what are the exact ways by which all these otherwise perfectly regulated mechanisms fail to protect the host? Next, we will analyze each of these steps and will try to understand what makes things go amiss.
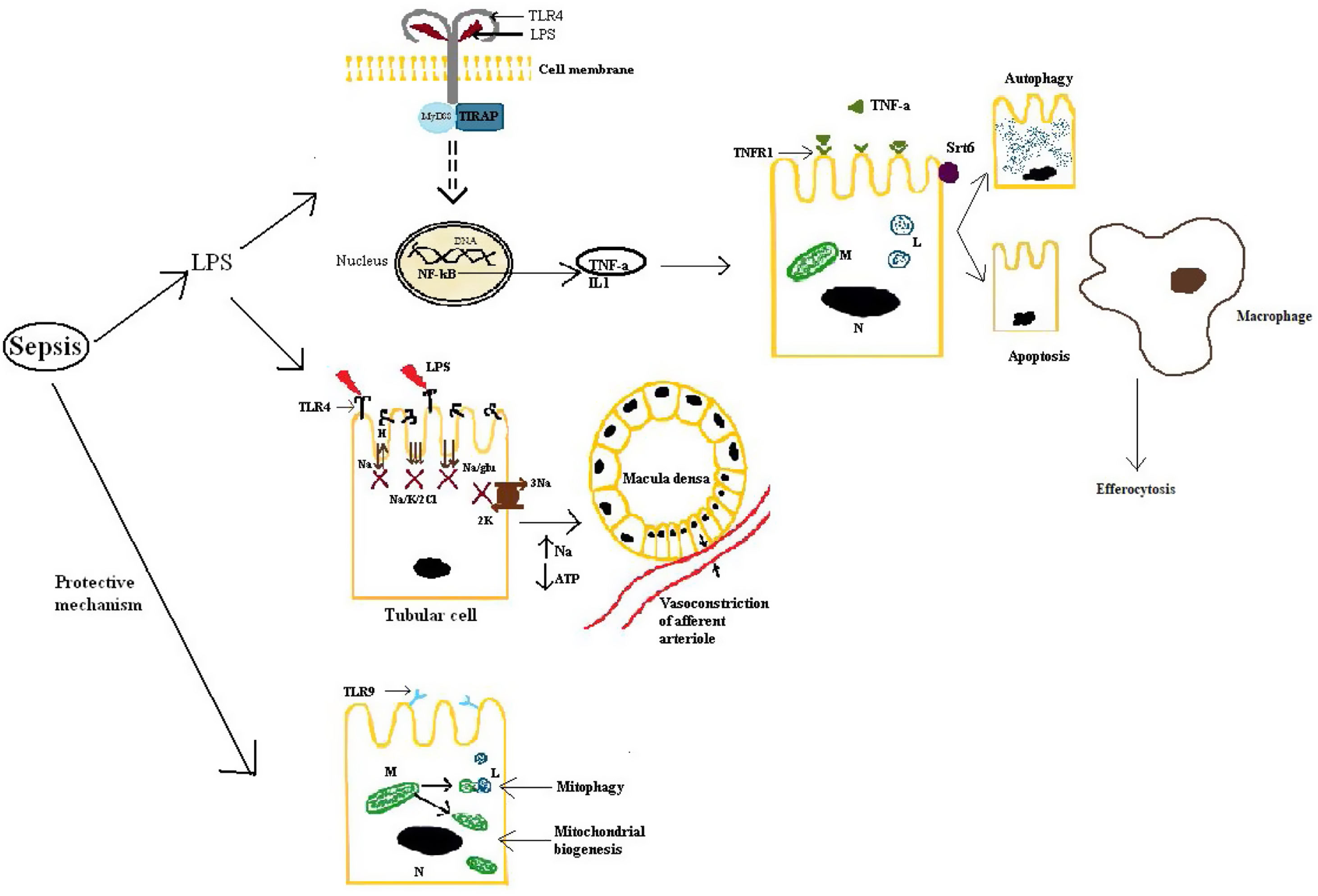
First of all, septic AKI is mediated by a response to DAMPs and PAMPs via toll-like receptors (TLRs), NOD-like receptors (NLRs) (NOD-like receptor) and RIG-I-like receptors (RLRs).29 TLR4 is the main receptor that binds LPS, leading to the release of TNF-α and IL-1 and activating many intracellular signals via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB).23 TLR4 is mainly expressed, but not only, on tubulocytes, the result being an alteration in the tubular ion transport, which decreases as soon as 9h after the induction of sepsis in animal models.30 This has two outcomes: one is that the energy requirement of the tubulocytes will decrease by 70%, as this is the cost of ion transport22 and the other is an increase in the load of sodium and chloride at macula densa. The overall effect is an enhancement of the tubulo-glomerular feedback leading to a decrease in the GFR, in the initial phases acting like a protecting mechanism, to lower the quantity of PAMPs reaching the renal tubules.31 Also, TLR2 and TLR4 increase in the monocytes of the septic patients and in the macrophages from the liver and spleen, the reason why they could become targets for sepsis-induced organ injury.31 Of all the cytokines involved via the TLR pathway, TNF-α has a very important role, especially in gram-negative induced sepsis in which endotoxins facilitate the release of it from mesangial cells.32,33 This will result in the upregulation of TNFR-1 (receptor 1 of TNF), which mediates apoptosis of the tubular cells.23 In addition, some authors argue that an elevated level of TNFR is an independent predictive factor for AKI and mortality.28 However, the role of apoptosis is debatable, at least in human beings. Morrell et al. evaluated many of the studies that conferred a central role to tubular cells apoptosis and concluded, based on histopathological research, that even though focal tubular injury is frequent (78%), the majority of tubular cells are normal, with no sign of apoptosis.22,23 To emphasize even more this fact it has to be mentioned that the conclusions of the majority of the studies which placed apoptosis in the central pathway of septic-AKI were based on observations on animal models and also, in clinical practice, therapies that inhibit caspase activation (Fas and caspase expression lead, at least in part, to apoptosis) lack significant benefit.23
Secondly, from an intracellular viewpoint, a few aspects have a very important role in protecting the tubular cells from inflammatory injury. Two of these are mitophagy and mitochondrial biogenesis, activated on the TLR9 pathway as a natural response during sepsis, an insufficient activation leading to worse outcomes in critically-ill patients, as they will not have enough cellular energy sources and also predisposing the tubulocytes to apoptosis.22,34 Another key aspect in renal protection is conferred by autophagy stimulated by sirtuins, especially sirtuin 6, and efferocytosis, by which inflammation is limited.35,36 Moreover, the decline of autophagy contributes to proximal tubular injury and dysfunction in sepsis.30 Another important aspect is that during septic AKI, the cell cycle stops in the G1–S phases to prevent the replication of damaged DNA.22 This is also the mechanism through which later renal cancer can develop.16
For an easier understanding, these aspects are also described in Fig. 2.

Kidney response to septic injury. LPS: lipopolysaccharide; TLR4: toll-like receptor 4; TLR9: toll-like receptor 9; MyD88, TIRAP: adapter proteins used by TLRs; DNA: deoxyribonucleic acid; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; TNF-α: tumor necrosis factor alpha; TNFR-1: tumor necrosis factor receptor 1; IL-1: interleukin 1; M: mitochondria; L: lysosome; N: nucleus; Srt6: sirtuin 6; Na: sodium; H: hydrogen ion; K: potassium; Cl: chloride; glu: glucose; ATP: adenosine triphosphate.
In the human body there are approximately 3.8×1013 microorganisms, almost with a ratio of one-to-one to human cells.37 Of particular importance are the microorganisms that live in the intestine, as they form the colo-renal axis.38 In the following, we will detail the mechanisms of the link between microorganisms and AKI severity.
As was already mentioned, inflammation with its variety of cytokines plays a very important role in sepsis and it is the key through which the majority of adverse outcomes develop. Of great importance is the inflammation of the gut, because alongside with hypoperfusion leads to intestinal injury, damage to the intestinal barrier, bacterial and toxins translocation and, in the end, amplification of systemic inflammatory response, multi-system organ failure (MSOF) and death.39
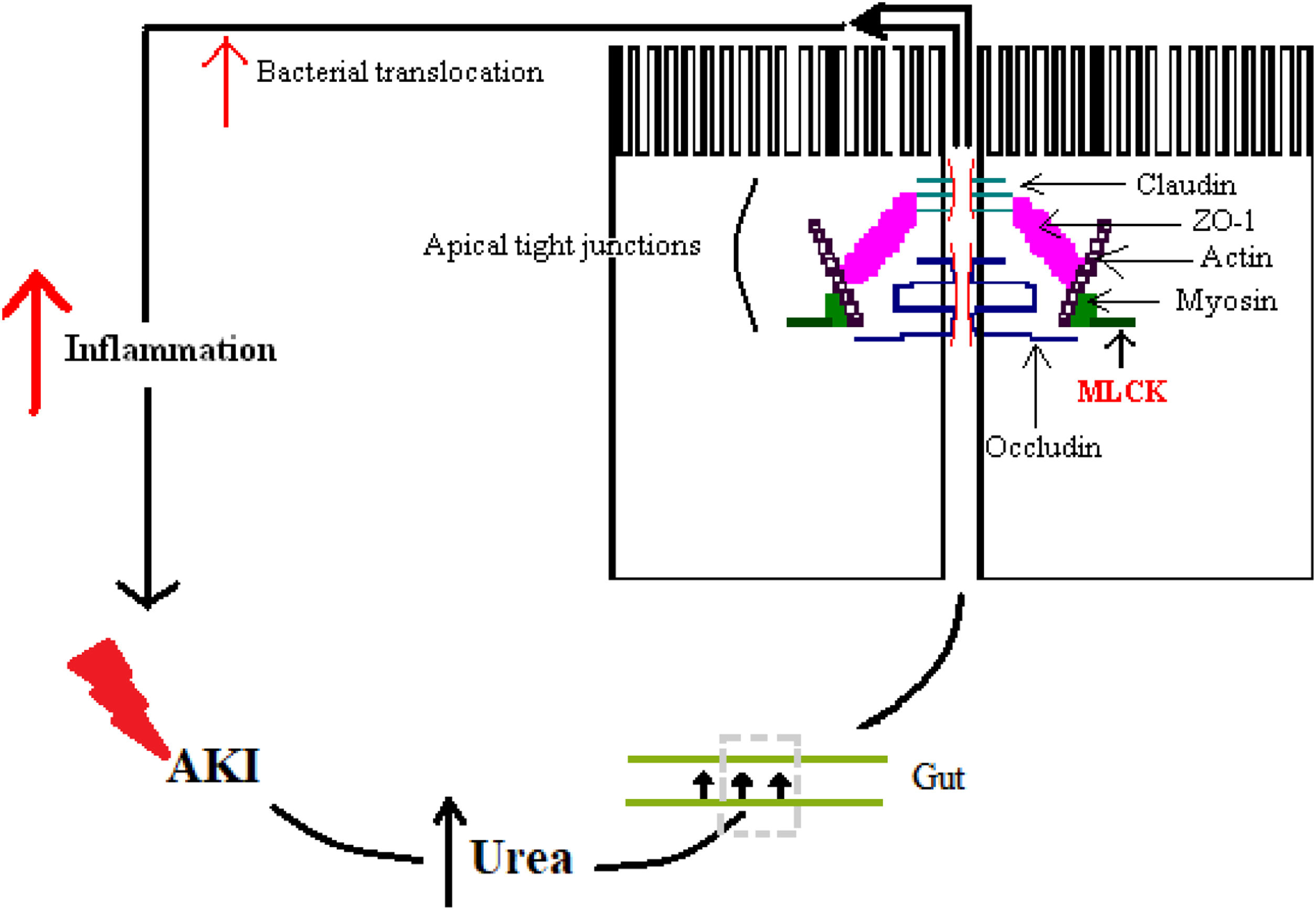
The alteration of the intestinal barrier develops in multiple ways: one way is by the accumulation of urea during AKI alongside with edema of the intestinal wall, which enables the diffusion of the urea in the intestinal lumen, where ammonia and caustic ammonium hydroxide form, the result being the disruption of the tight junctions.40 Another way is by the action of cytokines, which alter junctional proteins like zonula occludens 1 (ZO-1), claudins and occludins.41 A particular enzyme plays a crucial role and that is myosin light chain kinase (MLCK). Phosphorylation of this enzyme causes the contraction and opening of tight apical junctions of the intestinal barrier and, furthermore, its activation is associated with increased levels of other inflammatory cytokines like IL-6, TNF-α and IL-1β.41,42 Finally, there is also a particular cytokine in sepsis whose action further enhances the intestinal response and that is IL-17. It was shown that in AKI its release from intestinal Paneth cells is increased and it mediates hepatic and intestinal injury.43 The overall result is increased intestinal permeability, with the maintenance of a vicious circle of inflammation.41
Fig. 3 illustrates some of these alterations.

The gut microbiome has a major role in balancing all these effects through a specific type of fatty acids, respectively short chain fatty acids (SCFA), represented by acetate, propionate and butyrate, which are the end-products of dietary fiber fermentation by the microbiome.41 They have several roles, but two are well-described and constitute the link between the gut and the kidney. The first role is the capacity of modulating the immune response in the gut by activation of regulatory T cells (Tregs) and by enhancing the intestinal epithelial barrier, especially by butyrate, by promoting cellular proliferation, immune tolerance and providing energy for the colonocytes.41,43 The second role is kidney related. There are two receptors for SCFA: olfactory receptor 78 in the juxtaglomerular apparatus and one coupled with G proteins on endothelial cells.44 SCFA have hemodynamic effects via both receptors, modulating arterial pressure and ameliorating kidney dysfunction and GFR during septic AKI, especially acetate.41,44 Moreover, they improve the effect of hypoxia in renal epithelial cells by improving mitochondrial biogenesis.41 On the other hand, administration of acetate in long term can have adverse outcomes, in literature being described an increased risk of ureteritis and hydronephrosis mediated by T-cells.45
During septic AKI, 90% of anaerobic flora is lost and studies argue that the dysbiosis created is both a consequence of AKI and also a determinant of post-AKI severity by all the metabolical and immunological alterations induced.43 Consequently, targeting and protecting the microbiome could be of great importance and hence in the literature have been made efforts to study and understand how can dysbiosis be prevented or treated.41 First of all, it has to be mentioned that dysbiosis leads in AKI to an increase in Escherichia and Enterobacter and a decrease in Lactobacillus, Ruminococcaceae, Faecalibacterium and Lachnospiraceae, and to an even sharper decrease in SCFA as measured in feces.43 In addition, the number of neutrophils and macrophages in the colon increase, with the polarization of macrophages to the M1 pro-inflammatory subtype, leading to intestinal inflammation and leaky colon, and hence the facilitation of bacterial translocation and the aggravation of systemic immune response.41,43 On the other hand, during the recovery phase in AKI there is an increased number of Lactobacilli and butyrate and a decrease in the number of neutrophils, with the maintenance of the level of macrophages.43
All of these observations led to efforts to counteract the effects of dysbiosis and the most used way was by administration of broad-spectrum antibiotics for more or less time or selective digestive decontamination with non-absorbable antibiotics or antiseptics.38,43,46 The results were very impressive, with benefits for both the gut and the kidney: in the gut, the inflammation of the colon decreased, claudins 1 and 2 were restored, and, thus, the intestinal barrier, Th17 and Th1 lymphocytes also decreased and in the kidney, the tubular injury was minimized and GFR started to improve.41,43 Interestingly, macrophages started to polarize to M2 anti-inflammatory, which also facilitates tissue repair, both in the gut and the kidney after antibiotics.43 Most probably, all these effects are due to the modulation of the microbiome by the antibiotics, not to the elimination of the entire flora.38 Another way to counteract dysbiosis is by using pre- and probiotics like Lactobacillus spp. or Bifidobacterium spp. which in a study lowered the mortality in sepsis,41 and also SCFA, especially acetate when it comes to the amelioration of kidney function, but with caution, because, as we already mentioned, it can have deleterious effects on the long term.38,41 Finally, therapeutic strategies that limit the activation of monocytes/macrophages could be other therapies for the prevention of septic AKI.41
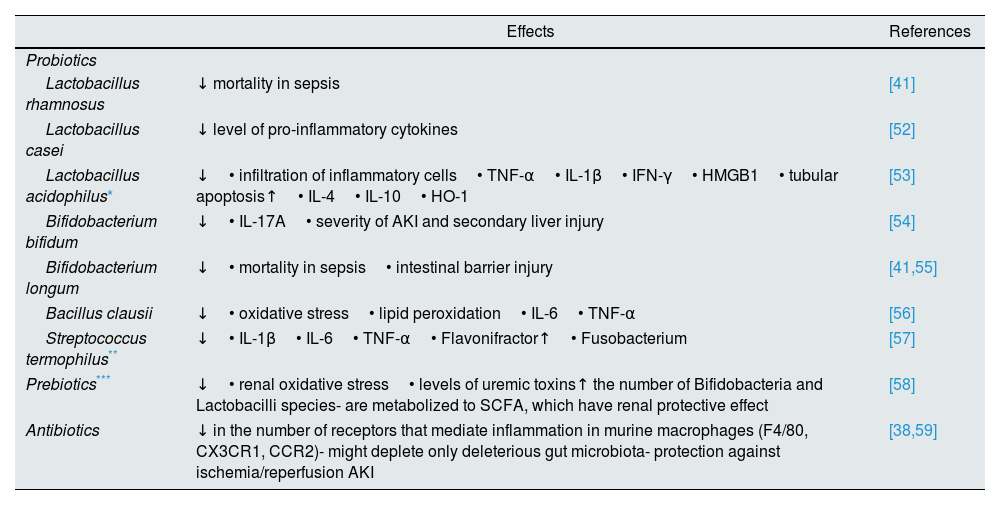
Table 1 presents a more detailed list of the strategies that interfere with intestinal microbiota.
Strategies that interfere with intestinal microbiome.
| Effects | References | |
|---|---|---|
| Probiotics | ||
| Lactobacillus rhamnosus | ↓ mortality in sepsis | [41] |
| Lactobacillus casei | ↓ level of pro-inflammatory cytokines | [52] |
| Lactobacillus acidophilus* | ↓• infiltration of inflammatory cells• TNF-α• IL-1β• IFN-γ• HMGB1• tubular apoptosis↑• IL-4• IL-10• HO-1 | [53] |
| Bifidobacterium bifidum | ↓• IL-17A• severity of AKI and secondary liver injury | [54] |
| Bifidobacterium longum | ↓• mortality in sepsis• intestinal barrier injury | [41,55] |
| Bacillus clausii | ↓• oxidative stress• lipid peroxidation• IL-6• TNF-α | [56] |
| Streptococcus termophilus** | ↓• IL-1β• IL-6• TNF-α• Flavonifractor↑• Fusobacterium | [57] |
| Prebiotics*** | ↓• renal oxidative stress• levels of uremic toxins↑ the number of Bifidobacteria and Lactobacilli species- are metabolized to SCFA, which have renal protective effect | [58] |
| Antibiotics | ↓ in the number of receptors that mediate inflammation in murine macrophages (F4/80, CX3CR1, CCR2)- might deplete only deleterious gut microbiota- protection against ischemia/reperfusion AKI | [38,59] |
IL: interleukin; TNF-α: tumor necrosis factor α; HMGB1: high-mobility group box 1; HO-1: heme oxygenase 1; AKI: acute kidney injury; SCFA: short chain fatty acids.; ↓: decrease; ↑: increase.
A lot of studies have looked for prediction factors in septic-AKI, especially because serum creatinine can be lowered in sepsis and hence is an imperfect estimator of renal function.4 Two markers involved in G1–S cell cycle arrest are claimed to be “the most sensitive and specific markers to predict the risk of development of AKI in critically ill patients”, respectively the tissue inhibitor of metalloproteinases 2 (TIMP2) and insulin-like growth factor-binding protein 7 (IGFBP7).22
Other studies consistently claimed neutrophil gelatinase-associated lipocalin (NGAL) as a good marker, but maybe even a more important aspect of this protein is the fact that it is not only a prediction factor but maybe a protective one too.28 This is explained by the fact that in septic AKI patients there are increased levels of NGAL and hepcidin, which suggests that disturbed iron homeostasis might be an important mechanism in this disease.4
Another important prognostic factor is IL-18, whose excretion is more increased in septic AKI than in other forms of kidney injury and it is also claimed that a raised level of IL-18 can predict the deterioration of kidney function with approximately 24–48h before clinical significant AKI. Also, as was already mentioned above, increased levels of TNFR were considered an independent predictor of AKI and mortality.28
However, maybe the best well-known prognostic factor and also the most available one worldwide to be determined by laboratory kit is serum lactate, as a measure of hypoperfusion during sepsis. The target with therapies is to maintain its level under 2mmol.5
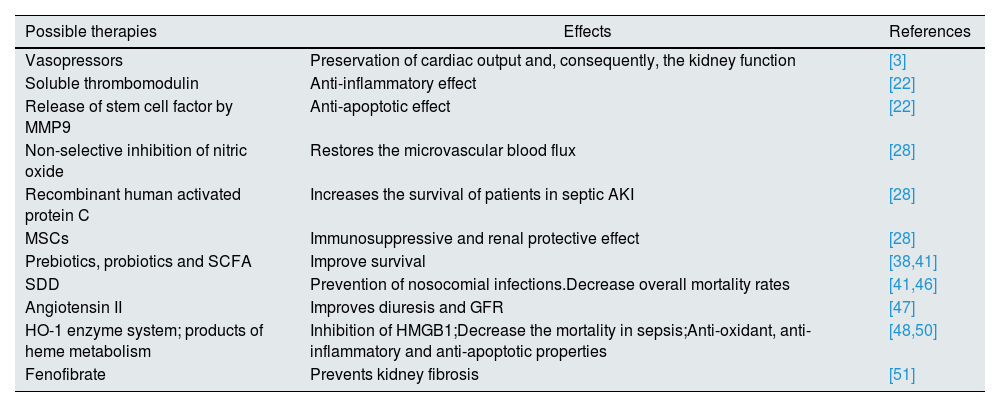
Possible therapiesWhen it comes to therapies for septic AKI, things have not improved significantly as only a few molecules reached clinical trials and provided benefits. One of these molecules is recombinant human activated protein C which increased the survival of patients with septic AKI.28 Another promising agent is represented by mesenchymal stem cells (MSCs) due to their immunosuppressive and renal protective effect by stimulating the proliferation and differentiation of tubular cells into mature cells.28 Also, in clinical trials, the infusion of angiotensin II can improve diuresis and GFR, without major adverse effects.47
Also, a comprehensive systematic review and meta-analysis suggested that selective decontamination of the digestive tract using non-absorbable antibiotics can prevent nosocomial infections in critically ill patients and decrease overall mortality rates.41,46 In addition, the use of pre- and probiotics like Lactobacillus rhamnosus and Bifidobacterium longum, along with SCFA, especially butyrate for the intestinal epithelial barrier and acetate for kidney dysfunction, can improve survival, at least on murine models.38,41
An interesting proposed therapy has been the utilization of the stress-responsive heme oxygenase-1 (HO-1) enzyme system and the products of heme catabolism, including carbon monoxide, biliverdin, and bilirubin, which could suppress, at least in animal models, the high-mobility group box 1 (HMGB1).48,49 This protein is a nonhistone chromatin-associated protein that plays a pivotal role in hematopoietic malignancies.49 What is appealing about it is the fact that in mice deficient in HO-1, HMGB1 contributes to lethality in endotoxemia, but after the administration of carbon monoxide and biliverdin, there was a significant reduction in its level.28 Furthermore, animal studies have shown that HO-1 lowers the mortality in sepsis, suppresses the infiltration of neutrophils in rat liver during sepsis through inactivation of p38 mitogen-activated protein kinase (MAPK) and, in addition, that it has anti-oxidant, anti-inflammatory and anti-apoptotic properties.48,50
Other agents are listed only as possible therapeutic options, without a clear demonstration of their benefic effect in human septic AKI: soluble thrombomodulin, which may have an anti-inflammatory effect in AKI, the release of stem cell factor by matrix metalloproteinase 9 (MMP9) which have an anti-apoptotic effect via activation of c-kit, a tyrosine kinase receptor found on the surface of the majority of the cells and non-selective inhibition of nitric oxide, which can restore the microvascular flux and, thus, preserve the kidney function.22,28 Another option is, as was observed in animal models, the administration of fenofibrate, which restores fatty acid oxidation (FAO)and prevents kidney fibrosis.51
Returning to the guide, KDIGO affirms that it is not known what vasopressor agent works best in preventing/treating septic AKI, but most studies focused on norepinephrine, dopamine and vasopressin.3 However, it is argued that some vasopressors can preserve better the kidney function – vasopressin analogs versus catecholamines.3 Also, fenoldopan seems to be protective in AKI by having anti-inflammatory effects independent of vasodilatory action, but also it poses a risk for hypotension and thus ischemia, with the risks involved.3
Table 2 outlines these possibilities.
Possible therapies in septic AKI.
| Possible therapies | Effects | References |
|---|---|---|
| Vasopressors | Preservation of cardiac output and, consequently, the kidney function | [3] |
| Soluble thrombomodulin | Anti-inflammatory effect | [22] |
| Release of stem cell factor by MMP9 | Anti-apoptotic effect | [22] |
| Non-selective inhibition of nitric oxide | Restores the microvascular blood flux | [28] |
| Recombinant human activated protein C | Increases the survival of patients in septic AKI | [28] |
| MSCs | Immunosuppressive and renal protective effect | [28] |
| Prebiotics, probiotics and SCFA | Improve survival | [38,41] |
| SDD | Prevention of nosocomial infections.Decrease overall mortality rates | [41,46] |
| Angiotensin II | Improves diuresis and GFR | [47] |
| HO-1 enzyme system; products of heme metabolism | Inhibition of HMGB1;Decrease the mortality in sepsis;Anti-oxidant, anti-inflammatory and anti-apoptotic properties | [48,50] |
| Fenofibrate | Prevents kidney fibrosis | [51] |
AKI: acute kidney injury; GFR: glomerular filtration rate; MMP9: matrix metalloproteinase 9; MSCs: mesenchymal stem cells; SDD: selective decontamination of the digestive tract; SCFA: short chain fatty acids; HO-1: heme-oxygenase 1; HMGB1: high-mobility group box 1.
The answer to this question is hard to be given. We now know that septic AKI is a hyperemic form of AKI, with particular hemodynamic mechanisms, that can be modulated by using vasopressors and by non-selective inhibition of nitric oxide, the last part still being studied. Also, we know that the tubular cells adapt to the inflammatory injury by decreasing ion transport and by increasing mitophagy and mitochondrial biogenesis, which additionally enhance the tubulo-glomerular feedback and lowers, initially as a protective mechanism, the GFR. Furthermore, a multitude of cells interact with each other to eliminate the offending agent, often injuring the host, like leukocytes, thrombocytes and monocytes/macrophages, some studies arguing that by limiting the activity of the last ones we could prevent the development of septic AKI.
Last, but not least, there is a key role for the gut microbiome in septic AKI as dysbiosis worsens AKI and AKI worsens dysbiosis, with bacterial translocation and marked reduction of SCFA, which have many protective functions like modulation of immune response in the gut by activation of regulatory T cells and by enhancing the intestinal epithelial barrier, and also hemodynamic capacity by modulation of arterial pressure and amelioration of kidney dysfunction and GFR. In clinical practice, administration of pre- and probiotics and SCFAs led to increased overall survival and also increased kidney function. Still, further research is needed as to develop strict guidelines and to compare between different probiotics.
In the end, the best answer that can be given is that we still do not know and do not understand a lot of intimate mechanisms that respond to internal or external offenders and this is demonstrated by the relatively stable incidence of AKI worldwide, but with all the studies mentioned above we are convinced that in the near future new therapies will appear and we could change the prognosis of septic AKI for the better.
Conflict of interestNone declared.



