





La enfermedad renal crónica (ERC) tiene una alta incidencia mundial y una tendencia ascendente que afecta principalmente a personas de edad avanzada. Cuando la ERC está muy avanzada se requiere el uso de terapias renales sustitutivas para prolongar la vida (diálisis o trasplante renal) y, pese a que la diálisis mejora muchas complicaciones de la ERC, la enfermedad no revierte de manera completa. Estos pacientes presentan un aumento del estrés oxidativo, inflamación crónica y aumento de la liberación de vesículas extracelulares (VE), que provocan daño endotelial y el desarrollo de distintas enfermedades cardiovasculares (ECV). De hecho, los pacientes con ERC desarrollan de forma prematura enfermedades asociadas a una edad avanzada, como es el caso de las ECV. Las VE desempeñan un papel muy importante en el desarrollo de ECV en pacientes con ERC, ya que su número aumenta en el plasma y su contenido se modifica. Las VE de pacientes con ERC generan disfunción endotelial, senescencia y calcificación vascular. Además, los miRNA libres o transportados en las VE junto a otros componentes vehiculados en estas VE promueven disfunción endotelial, eventos trombóticos y calcificación vascular en los pacientes con ERC, entre otros efectos. En esta revisión se describen los factores clásicos y el papel de nuevos mecanismos que intervienen en el desarrollo de la ECV asociada a la ERC, con especial hincapié en el papel de las VE en el desarrollo de enfermedades cardiovasculares en un contexto de ERC. Además, se expone el papel de las VE como herramienta diagnóstica y como diana terapéutica, actuando sobre su liberación o contenido para intentar evitar el desarrollo de ECV en enfermos renales crónicos.
Chronic kidney disease (CKD) is a pathology with a high worldwide incidence and an upward trend affecting the elderly. When CKD is very advanced, the use of renal replacement therapies is required to prolong its life (dialysis or kidney transplantation). Although dialysis improves many complications of CKD, the disease does not reverse completely. These patients present an increase in oxidative stress, chronic inflammation and the release of extracellular vesicles (EVs), which cause endothelial damage and the development of different cardiovascular diseases (CVD). CKD patients develop premature diseases associated with advanced age, such as CVD. EVs play an essential role in developing CVD in patients with CKD since their number increases in plasma and their content is modified. The EVs of patients with CKD cause endothelial dysfunction, senescence and vascular calcification. In addition, miRNAs free or transported in EVs together with other components carried in these EVs promote endothelial dysfunction, thrombotic and vascular calcification in CKD, among other effects. This review describes the classic factors and focuses on the role of new mechanisms involved in the development of CVD associated with CKD, emphasizing the role of EVs in the development of cardiovascular pathologies in the context of CKD. Moreover, the review summarized the EVs’ role as diagnostic and therapeutic tools, acting on EV release or content to avoid the development of CVD in CKD patients.
La enfermedad renal crónica (ERC) se considera un problema de salud pública ya, que afecta aproximadamente a 850 millones de personas en el mundo, y es responsable de unos 2,4 millones de muertes anuales; de hecho, en países desarrollados, alrededor del 11% de la población desarrolla ERC1. En España, hay cerca de 7 millones de personas afectadas, con un notable crecimiento (en torno al 4-5% cada año) en la última década2,3. Este incremento parece estar relacionado con diferentes causas, entre las que destaca el envejecimiento de la población. Hoy en día alrededor del 7% de la población mundial tiene más de 65 años y este porcentaje es alrededor del 15% en países desarrollados. Concretamente en España, alrededor del 35% de la población tiene más de 75 años4. Otra posible causa es el aumento de la prevalencia de otros factores de riesgo importantes como son la obesidad, la hipertensión arterial, el tabaquismo o la diabetes mellitus. Esta última enfermedad es la causa más frecuente de ERC: causó el 26,54% de los nuevos casos de ERC5 en España en el año 2020 (fig. 1) y la mortalidad por ERC en el mundo es del 44,7%3.

Enfermedad renal crónica, enfermedades asociadas y terapias. La ERC es producida por enfermedades como diabetes mellitus, poliquistosis o hipertensión arterial, entre otras. Como consecuencia de la ERC se producen enfermedades relacionadas con el daño vascular y la ECV, que están mediadas por distintos factores. En la parte inferior se muestran las principales modalidades de tratamiento en pacientes con ERC.
El deterioro de la función renal en pacientes con ERC sucede de manera progresiva y silenciosa. Es frecuente que los síntomas de ERC no aparezcan hasta que la enfermedad está muy avanzada (enfermedad renal crónica avanzada), incluso hasta poco antes de que los pacientes requieran tratamiento renal sustitutivo6. El tratamiento renal sustitutivo mediante diálisis ayuda a prolongar la vida de muchos pacientes en etapa terminal, aunque también puede complicar su estado clínico, al empeorar la funcionalidad cardiovascular7,8. Existen 2modalidades de diálisis: la hemodiálisis (HD) y la diálisis peritoneal. Ambas técnicas consiguen eliminar parte de las toxinas urémicas acumuladas en la sangre. La terapia idónea para resolver la enfermedad es el trasplante renal, aunque para ello se debe encontrar un riñón de un donante compatible, lo cual puede llevar meses o incluso años, y, aun así, no es una solución definitiva. Un porcentaje importante de los pacientes quedan con un cierto grado de insuficiencia renal crónica y la vida media de un trasplante de riñón está por debajo de los 20 años9 (fig. 1).
En los pacientes con ERC es común observar distintas comorbilidades, especialmente enfermedades cardiovasculares (ECV), que son las que constituyen la principal causa de mortalidad7,8. El incremento de la mortalidad por ECV ocurre desde los primeros estadios de la ERC10 desencadenado por el daño endotelial generado por ella11 (fig. 1). Controlar las diferentes comorbilidades y los factores de riesgo que pueden acelerar la disminución de la función renal es esencial para frenar el desarrollo de la enfermedad, al igual que su detección precoz, que hoy en día sigue siendo un reto5.
El alto riesgo de desarrollar ECV en pacientes urémicos por ERC hace necesarios la identificación de nuevos biomarcadores para la predicción y diagnóstico de complicaciones cardiovasculares y el desarrollo de nuevas terapias para el tratamiento de estas enfermedades12. En los últimos años se han identificado diferentes elementos y mecanismos que pueden estar implicados en el desarrollo de la ERC o ECV de forma precoz, entre los que destacan las vesículas extracelulares (VE), los microRNA (miRNA) o el recuento de ciertas poblaciones inmunitarias11.
El objetivo de este trabajo es revisar el estado del conocimiento de los mecanismos que determinan la aparición de ECV en los pacientes con ERC. Se analizarán aspectos implicados en el envejecimiento acelerado de los pacientes con ERC y, en particular, en el desarrollo del daño vascular, incluyendo el papel de las VE como un importante efector asociado al desarrollo de enfermedades complejas en la ERC. La identificación precoz de biomarcadores implicados en estos procesos es de enorme interés para hallar nuevas dianas terapéuticas y para prevenir o evitar el desarrollo de la ERC y la ECV asociada.
El desarrollo de la enfermedad cardiovascular en pacientes con enfermedad renal crónicaExisten numerosas conexiones entre la ERC y ECV, de hecho, comparten factores de riesgo como la hipertensión arterial, presente en la mayoría de los enfermos renales, la diabetes mellitus y la dislipidemia5, lo que sugiere que el desarrollo de la ECV asociado a ERC tiene un origen multifactorial.
La pérdida de la función renal induce alteraciones metabólicas y bioquímicas. Una de las alteraciones más importantes es la acumulación de toxinas urémicas que, al unirse con proteínas, no se pueden eliminar en los tratamientos de diálisis debido a su elevado peso molecular y van a desencadenar ECV13. Además, estas toxinas van a producir lesiones vasculares, como fibrosis de la túnica íntima, arteriosclerosis hiperplásica (relacionada con la hipertensión arterial), la formación de placa aterosclerótica y calcificación vascular de la túnica media. Estas 2últimas manifestaciones se asocian a una mayor morbimortalidad en pacientes con ERC14. Además, la acumulación de toxinas urémicas en pacientes con ERC causa disfunción endotelial: aumento de la permeabilidad vascular por pérdida de las uniones célula-célula, un incremento del estrés oxidativo y activación de diversas rutas de señalización proinflamatorias y protrombóticas15.
La diálisis también puede contribuir a la aparición de ECV debido al uso de líquidos de diálisis, inmunosupresores, anticoagulantes…, entre otros16. Estas terapias están asociadas con un aumento del estrés oxidativo y la inflamación sistémica que, mantenidos en el tiempo, alteran la respuesta inmune en los pacientes renales. Asimismo, en este tipo de pacientes se produce activación plaquetaria, de la cascada del complemento y del sistema inmune. Todos estos factores van a afectar al endotelio y derivan en un daño de la vasculatura, paso previo y desencadenante de múltiples enfermedades vasculares asociadas a pacientes con ERC17.
Daño endotelial asociado a la enfermedad renal crónicaEl endotelio es algo más que una simple barrera entre el torrente sanguíneo y la pared vascular: es un elemento activo que participa en una gran cantidad de procesos, entre ellos, la regulación del tono vascular, el crecimiento y migración de la fibra muscular lisa subyacente y el mantenimiento de la estructura vascular18. En condiciones fisiológicas, las células endoteliales presentan una superficie antiadherente y anticoagulante, pero en respuesta a un daño, las moléculas que se expresan en su superficie pueden verse modificadas y, con ello, puede aumentar la capacidad de adhesión celular. Las plaquetas se unen a la superficie dañada, lo que supone el inicio del proceso de coagulación, con el consiguiente desarrollo de inflamación y trombosis, propiciando la aparición de accidentes cardiovasculares. Por tanto, el daño endotelial puede ser el evento inicial que desencadene la aparición de diferentes enfermedades cardiovasculares11 (fig. 2).

Daño vascular. El estrés oxidativo y la inflamación, entre otros factores, generan un deterioro endotelial que conlleva: aumento de la capacidad de adhesión que provoca trombosis, aumento de la liberación de VE y mayor extravasación leucocitaria, lo que favorece el desarrollo de enfermedades cardiovasculares. Figura elaborada con Biorender.
Existen diferentes mecanismos que participan en la degeneración y deterioro del endotelio. A continuación, se describen los más destacados en los pacientes con ERC.
Estrés oxidativoEl estrés oxidativo se define como la acumulación de moléculas oxidantes, principalmente especies reactivas de oxígeno (ROS), bien por el aumento de su producción o por una disminución de los mecanismos antioxidantes del organismo. Las ROS presentan una gran capacidad oxidante, por lo que pueden alterar diferentes moléculas (proteínas, lípidos, ácidos nucleicos…) de las células y generar daño en ellas19,20. En fases tempranas de la ERC se han observado altos niveles de estrés oxidativo y su correlación con la progresión de la enfermedad21,22. Además, los pacientes con ERC acumulan en su torrente sanguíneo toxinas urémicas, entre las que destacan el IS (indoxil sulfato), malondialdehído y dimetilarginina asimétrica, que empeoran la situación de estrés oxidativo23.
En general, el estrés oxidativo puede producirse por diversas causas (fig. 3). Los pacientes con ERC, principalmente aquellos con nefropatía diabética, presentan una disfunción mitocondrial que supone un aumento de la producción de ROS. En modelos animales y estudios in vitro se ha observado un aumento de la actividad de la enzima NADH oxidasa 4 en presencia de IS, lo que también supone un incremento en la producción de ROS23.

Las ROS también alteran estructuras celulares y vías metabólicas. Uno de los marcadores usados para medir el estrés oxidativo son los llamados AGE (advanced glycation end products o productos finales de glicación avanzada). Los AGE se unen a sus receptores RAGE (receptor system for AGE) y señalizan por la vía de las MAP cinasas (MAPK). La activación de las MAPK permite la internalización en el núcleo de la subunidad p65 del NF-κB (nuclear factor kappa B) y, como consecuencia final, el aumento de la liberación de citocinas y enzimas proinflamatorias, así como el aumento de la expresión de moléculas de adhesión24. Al mismo tiempo, las células inmunitarias proinflamatorias, como están activas, liberan componentes oxidantes, por lo que se genera un círculo vicioso que amplifica el daño oxidativo25.
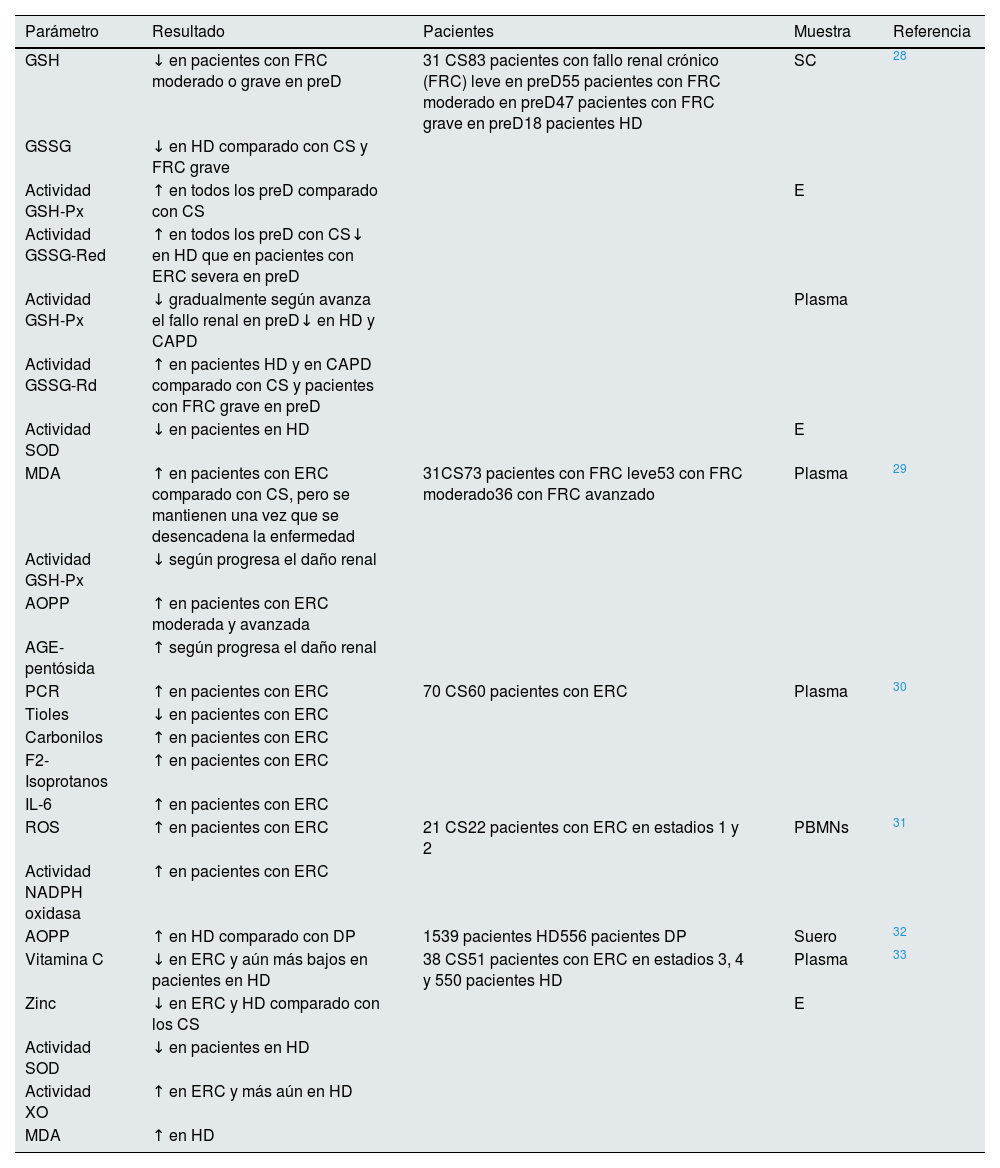
El aumento del estrés oxidativo daña el ADN. En concreto, la guanina puede oxidarse y transformarse en 8-hidroxi-2’-desoxiguanosina (8-OH-dG), otro marcador usado para medir el estrés oxidativo26. Este cambio de bases genera daño celular y se ha asociado con múltiples enfermedades crónicas y degenerativas, entre ellas, la ERC y los carcinomas27. Además, existen numerosos factores que incrementan de forma gradual en la ERC y muestran el declive de la función renal, por lo que se consideran como los causantes del aumento del estrés oxidativo (tabla 1).
Estrés oxidativo asociado a la ERC
| Parámetro | Resultado | Pacientes | Muestra | Referencia |
|---|---|---|---|---|
| GSH | ↓ en pacientes con FRC moderado o grave en preD | 31 CS83 pacientes con fallo renal crónico (FRC) leve en preD55 pacientes con FRC moderado en preD47 pacientes con FRC grave en preD18 pacientes HD | SC | 28 |
| GSSG | ↓ en HD comparado con CS y FRC grave | |||
| Actividad GSH-Px | ↑ en todos los preD comparado con CS | E | ||
| Actividad GSSG-Red | ↑ en todos los preD con CS↓ en HD que en pacientes con ERC severa en preD | |||
| Actividad GSH-Px | ↓ gradualmente según avanza el fallo renal en preD↓ en HD y CAPD | Plasma | ||
| Actividad GSSG-Rd | ↑ en pacientes HD y en CAPD comparado con CS y pacientes con FRC grave en preD | |||
| Actividad SOD | ↓ en pacientes en HD | E | ||
| MDA | ↑ en pacientes con ERC comparado con CS, pero se mantienen una vez que se desencadena la enfermedad | 31CS73 pacientes con FRC leve53 con FRC moderado36 con FRC avanzado | Plasma | 29 |
| Actividad GSH-Px | ↓ según progresa el daño renal | |||
| AOPP | ↑ en pacientes con ERC moderada y avanzada | |||
| AGE-pentósida | ↑ según progresa el daño renal | |||
| PCR | ↑ en pacientes con ERC | 70 CS60 pacientes con ERC | Plasma | 30 |
| Tioles | ↓ en pacientes con ERC | |||
| Carbonilos | ↑ en pacientes con ERC | |||
| F2-Isoprotanos | ↑ en pacientes con ERC | |||
| IL-6 | ↑ en pacientes con ERC | |||
| ROS | ↑ en pacientes con ERC | 21 CS22 pacientes con ERC en estadios 1 y 2 | PBMNs | 31 |
| Actividad NADPH oxidasa | ↑ en pacientes con ERC | |||
| AOPP | ↑ en HD comparado con DP | 1539 pacientes HD556 pacientes DP | Suero | 32 |
| Vitamina C | ↓ en ERC y aún más bajos en pacientes en HD | 38 CS51 pacientes con ERC en estadios 3, 4 y 550 pacientes HD | Plasma | 33 |
| Zinc | ↓ en ERC y HD comparado con los CS | E | ||
| Actividad SOD | ↓ en pacientes en HD | |||
| Actividad XO | ↑ en ERC y más aún en HD | |||
| MDA | ↑ en HD |
AGE: productos de glicación avanzada; AOPP: productos proteicos de glicación avanzada; CAPD: diálisis peritoneal continua ambulatoria; CS: controles sanos; DP: diálisis peritoneal; E: eritrocitos; ERC: enfermedad renal crónica; FRC: fallo renal crónico; GSH: glutatión; GSH-Px: glutatión peroxidasa; GSSG: glutatión disulfuro; GSSG-Red: glutatión reductasa; HD: hemodiálisis; IL-6: interleucina 6; MDA: malondialdehído; PBMN: células mononucleares de sangre periférica; PCR: proteína C reactiva; preD: prediálisis; ROS: especies reactivas del oxígeno; SC: sangre completa; SOD: superóxido dismutasa; XO: xantina oxidasa.
La hiperfosfatemia, presente en muchos pacientes con ERC, también tiene un papel importante en el deterioro vascular y está relacionada con el estrés oxidativo. Experimentos realizados in vitro con células endoteliales mostraron que las altas concentraciones de fosfato generaban un aumento del estrés oxidativo y una reducción del óxido nítrico (NO). En condiciones patológicas tiene lugar un descenso en la síntesis de NO como consecuencia de la inhibición de la fosforilación de la enzima óxido nítrico sintasa endotelial (eNOS)34. El NO es liberado por las células endoteliales para su relajación en condiciones fisiológicas y así evitar la rigidez de las arterias, por lo que un descenso de su producción favorece el deterioro endotelial. Estos resultados se han visto corroborados por experimentos realizados en ratones, tanto sanos como con ERC: las dietas altas en fosfato promueven la inflamación y la disfunción endotelial, además de una pérdida de permeabilidad entre las células endoteliales35.
Numerosos estudios muestran como el estrés oxidativo se encuentra relacionado directamente con factores proinflamatorios36-38. Los pacientes en HD pueden presentar un incremento de hasta 30 o 40 veces de la concentración fisiológica de acetato tras la sesión de diálisis, ya que es el ácido más frecuente utilizado en los líquidos de diálisis. Este aumento del acetato en sangre está asociado con un aumento en el estrés oxidativo y también con el incremento de algunas citocinas proinflamatorias, como las interleucinas 1 y 6 (IL-1 e IL-6) y con la síntesis de NO36-38. Si, además, hay una alta concentración de magnesio en el líquido de diálisis (1,25 o 2mM), se incrementa la producción in vitro de ROS y malondialdehído, aunque este incremento de magnesio se ha visto que protege contra el aumento de estrés oxidativo cuando en lugar de utilizarse acetato se usa citrato, como en los líquidos de diálisis (citrato solo o con una mezcla de citrato y pequeña cantidad de acetato)16. El aumento de la concentración de glutatión oxidado como resultado de la HD hace que la técnica de diálisis se considere un factor de riesgo asociado a un incremento del estrés oxidativo39.
Por tanto, en los pacientes con ERC, la uremia desencadena un incremento del estrés oxidativo que afecta en especial a las células endoteliales debido a su contacto directo con la sangre. A su vez, este estrés oxidativo genera alteraciones que van a propiciar el desarrollo de procesos inflamatorios, de tal forma que la coexistencia de la inflamación y el estrés oxidativo van a desencadenar y potenciar la disfunción endotelial en pacientes con ERC.
Mediadores inflamatorios y respuesta inmune en ERCLa inflamación desempeña un papel importante en el aumento del riesgo de accidentes cardiovasculares en los pacientes con ERC. Se ha descrito que los procesos inflamatorios se desencadenan como consecuencia de factores como la hipertensión arterial, la edad avanzada, la diabetes mellitus, la obesidad, la hiperuricemia, la dislipidemia, el tabaquismo, el sexo masculino y los antecedentes familiares de ECV40. Además, tanto la acumulación de toxinas urémicas como el tratamiento dialítico para depurarlas promueven el desarrollo de la inflamación40,41.
Los pacientes con ERC presentan inflamación crónica causada por la uremia y por las técnicas de diálisis42. Estos procesos inflamatorios en pacientes en diálisis son más frecuentes en estadios avanzados de la enfermedad. En particular, la continua irritación del peritoneo producida durante la diálisis peritoneal conduce a la activación de genes relacionados con la inmunidad adaptativa y la respuesta de linfocitos Th243. En el caso de la HD, el uso de membranas no biocompatibles o la posible contaminación del líquido de diálisis, entre otros factores44, van a generar la activación de monocitos, que liberan citocinas proinflamatorias. No obstante, la presencia de inflamación en pacientes en prediálisis indica que la activación de monocitos no es la principal causante de la inflamación45-47.
Al inicio de la ERC, los pacientes presentan una inflamación sistémica de baja intensidad que, al verse prolongada en el tiempo, genera un deterioro de la condición general del organismo y favorece la aparición de enfermedades secundarias, como la ECV48. En concreto, se observa una leve elevación de marcadores inflamatorios, como la proteína C reactiva, y de algunas citocinas, como la IL-6 y el factor de necrosis tumoral α (TNF-α)49. Incluso se puede considerar que la proteína C reactiva es un marcador de mortalidad, ya que se ha observado que con menores niveles de esta proteína existe un menor riesgo de mortalidad en pacientes en HD50. Por otra parte, IL-6, IL-1 y TNF-α están directamente relacionadas con la gravedad de la ERC. Se ha descrito la IL-6 como marcador predictivo de arteriosclerosis, ya que contribuye a la generación de la placa de ateroma y a su desarrollo mediante diversos mecanismos51. Se desconocen las vías de señalización concretas que utiliza la IL-6 en el desarrollo de arteriosclerosis. En modelos animales se ha observado que, al unirse la IL-6 a su receptor IL6-R, se activa la vía de señalización IL-6 trans, que desencadena un proceso inflamatorio crónico y favorece la formación de la placa de ateroma y, con ello, el desarrollo de enfermedades cardiovasculares52. El IS es capaz de unirse al receptor de aril-hidrocarburo (aryl hydrocarbon receptor, AhR) de los monocitos para que secreten TNF-α. El TNF-α va a interaccionar con las células endoteliales aumentando la expresión de la quimiocina CX3CL1 (también conocida como fractalcina), cuyo receptor es CX3CR1, altamente expresado en los linfocitos T CD4+CD28–. Esta subpoblación de linfocitos T se encuentra elevada en pacientes con ERC y, si son estimulados a través del receptor de células T, pueden inducir apoptosis en las células endoteliales y acelerar la progresión de ECV53 (fig. 4).

Disfunción endotelial y calcificación vascular generada por la unión del IS al receptor AhR en monocitos. El aumento de TNF-α generado por el IS genera un aumento de la producción de la quimiocina CX3CL1 en las células endoteliales. Cuando CX3CL1 se une a su receptor en el linfocito T, y este tiene su receptor de células T activo, va a generar la entrada en apoptosis de la célula endotelial. Además, TNF-α promueve osteólisis y liberación de calcio y fosfato, que favorecen la calcificación vascular.
AhR: receptor de aril-hidrocarburo; BMP2: proteína morfogénica ósea 2; IS: indoxil sulfato; MSX2: Msh homrbox protein 2; Runx2: runt-related transcription factor 2; TCR: receptor de célula T; TNF-α: factor de necrosis tumoral alfa; TNFR: receptor de TNF.
El TNF-α es uno de los principales activadores de RANKL (ligando del receptor activador para el factor nuclear κB). De esta forma, el TNF- α en el tejido óseo de los pacientes con ERC puede activar a los osteoclastos, alterar la estructura ósea y aumentar el riesgo de fracturas, lo que explica la alta frecuencia de fracturas en pacientes en HD54. Además, la activación de osteoclastos desencadena un aumento de los niveles de calcio y fosfato en sangre que facilita los procesos de mineralización de las células vasculares. El TNF-α potencia este proceso de mineralización, ya que permite la translocación de NF-κB, aumenta la liberación de VE (que en enfermos renales contienen altos niveles de calcio) e incrementa el nivel de la proteína morfogénica ósea 2, induciendo la diferenciación osteogénica y calcificación de las células musculares lisas vasculares (CMLV)55.
En pacientes con ERC, las células endoteliales activadas expresan también una mayor cantidad de VEGF (vascular endothelial growth factor) que, a su vez, promueve la angiogénesis y la remodelación de los vasos para contrarrestar la hipoxia de los tejidos renales. Igualmente, en pacientes que presentan proteinuria debido a un descenso de la filtración glomerular se ha visto que las células endoteliales del glomérulo incrementan la liberación de VEGF56,57.
Vesículas extracelularesLa acumulación de toxinas urémicas se ha asociado a un daño endotelial que tiene como resultado, entre otros efectos, un incremento de la liberación de VE58,59. Las VE sirven como sistema de señalización local y sistémico, y están involucradas en la función y la homeostasis del organismo60. En general, son muy diversas y pueden encontrarse en todos los fluidos corporales, incluyendo la leche materna y la orina61. Las VE son producidas por la mayoría de las células, incluidas las células B, las células T, los monocitos y las células endoteliales. La formación de estas VE es parte de la función celular normal; no obstante, aumentan en condiciones de estrés celular, apoptosis o viabilidad celular alterada, como es el caso de la ERC62.
Las VE se pueden clasificar según su origen, composición y tamaño en: a) exosomas (40-120nm), producto de la exocitosis de cuerpos multivesiculares; b) microvesículas (MV) (50-1.000nm), también conocidas como micropartículas, que surgen de evaginaciones de la membrana plasmática y c) los cuerpos apoptóticos (1-5μm), que se forman en los estadios finales de la apoptosis11,40. A efectos prácticos, debido a la dificultad para aislarlas y a la falta de un protocolo que permita obtener un único tipo de VE, resulta casi imposible distinguir un tipo de otro, especialmente entre los 2primeros. Por ello, en la actualidad se tiende a hablar de ellas de forma generalizada como VE, sin hacer distinción entre clases.
En los riñones, las VE han sido estrechamente vinculadas a procesos patológicos, como la inflamación, fibrosis, trombosis y supresión inmunológica63. Las MV y los exosomas pueden desempeñar activamente papeles patológicos en el desarrollo y la progresión de las ECV porque actúan como portadores de diversos miRNA40. Además, están involucradas en el desarrollo de la calcificación vascular como consecuencia del estado de envejecimiento vascular acelerado64. De este modo, se han propuesto las VE y su contenido como dianas terapéuticas: podrían ser inhibidas para revertir o tratar la progresión de la enfermedad renal. Además, las VE pueden participar en varios procesos de reparación tisular y modulación inmunológica, ya que podrían utilizarse directamente como agentes terapéuticos en la medicina regenerativa y el tratamiento de enfermedades autoinmunes.
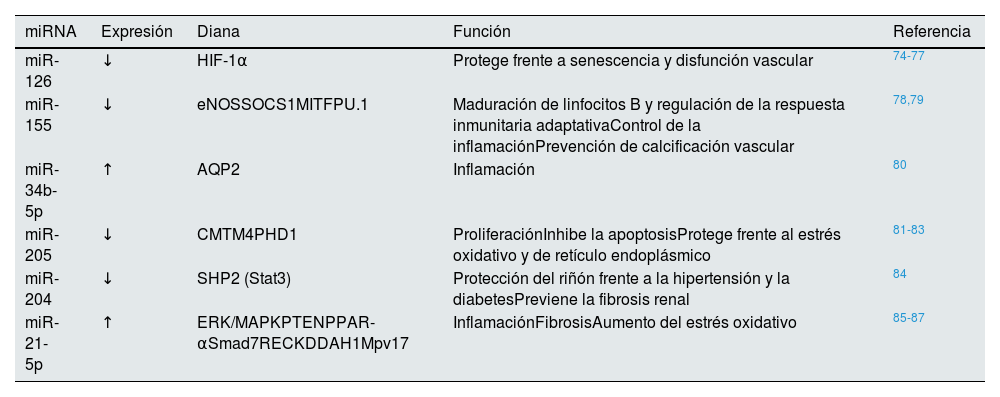
MicroRNALas VE transportan proteínas, lípidos y ácidos nucleicos, entre los que destacan los miRNA, aunque estos últimos también pueden ser transportados de forma libre por la sangre65. Los miRNA son ARN de pequeño tamaño (aproximadamente 22 nucleótidos de longitud), no codificantes, cuya función principal es la regulación de la expresión de proteínas66. Los miRNA tienen 2formas de actuación: pueden inhibir parcialmente la expresión de una proteína al unirse a la región 3’ UTR del transcrito, donde inician la degradación del ARN mensajero e inhiben su traducción; o pueden interaccionar con el promotor, favoreciendo el reclutamiento de la maquinaria de transcripción del gen y, por tanto, aumentando la expresión de la proteína67,68. La mayoría de los miRNA están presentes dentro de la célula, aunque también pueden encontrarse en fluidos corporales, como la sangre69,70. Para evitar su degradación por enzimas con actividad RNasa, se postula que estos miRNA extracelulares se encuentren empaquetados dentro de VE, fundamentalmente exosomas y MV69,71,72. Los miRNA son altamente específicos según el tipo de tejido y de célula, incluso, de la etapa de desarrollo y participan en una gran cantidad de procesos fisiopatológicos, incluyendo la inflamación. En el entorno vascular, los miRNA actúan como moléculas reguladoras en las células endoteliales, las CMLV, plaquetas y otras células de la sangre73. En la tabla 2 se incluyen los miRNA más relevantes cuya expresión se ha visto modificada en la ERC.
miRNA que participan en enfermedades vasculares cuya expresión está modificada en pacientes con ERC
| miRNA | Expresión | Diana | Función | Referencia |
|---|---|---|---|---|
| miR-126 | ↓ | HIF-1α | Protege frente a senescencia y disfunción vascular | 74-77 |
| miR-155 | ↓ | eNOSSOCS1MITFPU.1 | Maduración de linfocitos B y regulación de la respuesta inmunitaria adaptativaControl de la inflamaciónPrevención de calcificación vascular | 78,79 |
| miR-34b-5p | ↑ | AQP2 | Inflamación | 80 |
| miR-205 | ↓ | CMTM4PHD1 | ProliferaciónInhibe la apoptosisProtege frente al estrés oxidativo y de retículo endoplásmico | 81-83 |
| miR-204 | ↓ | SHP2 (Stat3) | Protección del riñón frente a la hipertensión y la diabetesPreviene la fibrosis renal | 84 |
| miR-21-5p | ↑ | ERK/MAPKPTENPPAR-αSmad7RECKDDAH1Mpv17 | InflamaciónFibrosisAumento del estrés oxidativo | 85-87 |
ARNT: transportador nuclear del receptor de aril-hidrocarburo; ATE: aterosclerosis; BCL2: linfoma de célula B2; CMLV: células musculares lisas vasculares. eNOS: sintasa de óxido nítrico endotelial; ERC: enfermedad renal crónica; HIF-1α: factor inducible por hipoxia 1α; KLF4/5: factor de transcripción Kruppel-like 4/5; Mϕ: macrófagos; PDCD4: proteína de muerte celular programada 4; PTEN: homólogo de tensina y fosfatasa; TNF-α: factor de necrosis tumoral α; VEGF-A: factor de crecimiento endotelial vascular-A.
El papel del miR-21 en la ERC es relevante. Se ha observado que miR-21-5p está sobreexpresado en pacientes con ERC y que sus niveles se correlacionan negativamente con la tasa de filtración glomerular86. Además, miR-21-5p inhibe a Smad7, un inhibidor de la ruta del TFG-β1/Smad3, por lo que esta ruta se ve potenciada, lo que genera inflamación y fibrosis renal en estos pacientes. Experimentos realizados en animales también muestran que miR-21 también es un inhibidor de la proteína Mpv17, encargada de reducir la producción de ROS en la mitocondria, por lo que el aumento de este miRNA en el riñón genera un aumento del estrés oxidativo87.
Otro miRNA importante es miR-155. Su ausencia o disminución en las VE derivadas de monocitos en un modelo de uremia genera alteraciones inmunitarias. Esto se debe a que miR-155 está implicado principalmente en la maduración de los linfocitos B, la regulación de la respuesta inmunitaria adaptativa y el control de la inflamación, ya que se expresa en linfocitos B y T activados, monocitos, macrófagos y células dendríticas78. Estas alteraciones inmunitarias podrían relacionarse con la inflamación y el daño vascular observado en los pacientes con ERC, pues su expresión es muy importante a la hora de dar forma al transcriptoma de células mieloides y linfoides activas78. La reducción de miR-155 también se ha asociado con la osteoclastogénesis y el desarrollo de calcificación vascular79.
Envejecimiento y senescencia celular: mecanismo implicado en el daño endotelial y vascular en ERCEl envejecimiento se define como un declive funcional del organismo que suele llevar implícitos cambios morfológicos y fisiológicos que suponen una pérdida en las reservas homeostáticas88. Existen 2tipos de envejecimiento: el envejecimiento fisiológico y el prematuro o acelerado, asociado a enfermedades. El envejecimiento fisiológico es la consecuencia del paso del tiempo y, por tanto, está íntimamente relacionado con la edad. Cuando el envejecimiento aparece de forma precoz y viene acompañado con el desarrollo de enfermedades asociadas al envejecimiento, se puede identificar con el denominado envejecimiento prematuro asociado a enfermedades. De este modo, los enfermos renales presentan un envejecimiento acelerado, ya que muchos de ellos desarrollan de forma precoz enfermedades asociadas al envejecimiento, como son las ECV4,18,89. El envejecimiento prematuro en estos pacientes se puede asociar con la acumulación de células senescentes, que son aquellas que han perdido su capacidad de proliferación.
Se define la senescencia celular como un proceso que tiene lugar como consecuencia del daño celular ocasionado por múltiples causas (estrés oxidativo, citocinas proinflamatorias o VE), y cuyo resultado final es que las células pierden su capacidad de división, no mueren y contribuyen al desarrollo de enfermedades asociadas a la edad. Entre otros cambios, son células que presentan un aumento de ROS, acumulación del número de mitocondrias disfuncionales, menor condensación de la heterocromatina constitutiva con pérdida de lámina β y un aumento de la actividad de la enzima β-galactosidasa lisosomal88. Además, las células senescentes presentan un déficit en la síntesis de proteínas, así como un aumento de su degradación. Obviamente, el aumento de células senescentes en un tejido desencadena una disfunción tisular.
En pacientes con ERC, la senescencia celular se produce en respuesta al daño generado por la acción de las toxinas urémicas acumuladas durante el progreso de la enfermedad90,91. La senescencia celular acelerada atribuida a la uremia genera un aumento del estrés oxidativo que, a su vez, ocasiona un deterioro de las estructuras celulares. Como consecuencia, se producen señales intracelulares que hacen que la célula adquiera un fenotipo senescente48. En el ámbito sistémico, las células senescentes liberan factores proinflamatorios que, si persisten en el tiempo, generan un deterioro generalizado del organismo. Este deterioro favorece la aparición de procesos tumorales y enfermedades degenerativas y crónicas92.
Las células renales son las encargadas de producir la proteína klotho, enzima capaz de hidrolizar el esteroide β-glucorinida. Es una proteína con efecto antienvejecimiento, capaz de modular la senescencia inducida por estrés y la respuesta funcional. La disminución de los niveles intracelulares de klotho se ha asociado con una senescencia endotelial, mientras que la klotho exógena previene la senescencia celular inhibiendo el incremento de estrés oxidativo inducido por la uremia, así como inactivando NF-κB al inhibir su capacidad de unión al ADN. De este modo se estabiliza el complejo NFκB/IκB y se reduce la liberación de citocinas que participan en procesos inflamatorios. Al estar disminuidos los niveles de esta proteína debido a la uremia, se observa un aumento del estrés oxidativo y una mayor senescencia endotelial93,94. Para el mantenimiento de la integridad y la homeostasis de las células endoteliales se ha observado que la proteína α-klotho juega un papel fundamental. En concreto, las funciones que se le han atribuido a α-klotho son la supresión de la expresión de moléculas de adhesión, como ICAM y VCAM, la atenuación de la vía de señalización NF-κB95 y la prevención de la hiperpermeabilización y la apoptosis de las células endoteliales, al mediar la internalización del complejo formado por el receptor de potencial transitorio canónico 1 y el receptor del factor de crecimiento endotelial vascular 296. Los pacientes con ERC presentan una deficiencia de esta proteína, aunque en esta deficiencia pueden intervenir múltiples factores19. Este déficit podría explicar, al menos en parte, el mayor riesgo de ECV. Además, se ha demostrado que klotho, junto con el factor de crecimiento de fibroblastos 23, (FGF23) resulta esencial en el eje hueso-riñón, ya que klotho mantiene la homeostasis del fosfato, colaborando de alguna forma con este facror de crecimiento (posiblemente deben estar bajo una misma ruta de señalización inhibiendo la reabsorción de fosfato), ya que la degradación de dicho factor lleva a un empeoramiento de la función renal, con hiperfosfatemia94.
Por último, cabe destacar de nuevo que el daño endotelial va a ser el paso previo al desarrollo de diversas ECV. La ECV asociada a la ERC está en parte determinada por una rigidez de los vasos sanguíneos ocasionada por un estado de inflamación y una pérdida de equilibrio entre los sistemas proenvejecimiento y antienvejecimiento que, finalmente, desencadena un proceso de calcificación vascular. En el ámbito vascular, se observa que los pacientes con ERC presentan un tejido vascular envejecido, similar al encontrado en individuos sanos de edad avanzada64.
Fenotipo secreto en la senescencia celular en pacientes con enfermedad renal crónicaCuando las células alcanzan un estado de senescencia sufren cambios en su fenotipo. El fenotipo secretor que adquiere una célula cuando entra en senescencia se denomina senescence-associated secretory phenotype (SASP). El material secretado se compone principalmente de quimiocinas, citocinas proinflamatorias (IL-1, IL-6, TNF-α, factor de crecimiento transformante β o TFG-β) y proteasas, de tal forma que, cuando las células senescentes se acumulan, hay una gran liberación de estos compuestos que genera una inflamación persistente de bajo nivel. Para evitar esto, las citocinas y quimiocinas van a atraer a las células del sistema inmunitario, principalmente monocitos y macrófagos, que van a eliminar estas células senescentes91.
En individuos envejecidos se acumulan las células senescentes porque existe un mayor número de células que van a entrar en senescencia y porque su eliminación va a ser más lenta, ya que su sistema inmune también es senescente. Como las células senescentes liberan factores proinflamatorios, el individuo envejecido presenta una inflamación crónica de bajo nivel97. El factor de transcripción NF-κB, la proteína cinasa activada por mitógenos p38 (p38 MAPK) y el inflamasoma son importantes efectores moleculares en el desarrollo de este fenotipo. Además, IL-1 y TGF-β generan un ciclo de retroalimentación positiva autocrina que propicia la situación proinflamatoria90,91. Y a todo esto se suman los resultados obtenidos en un modelo múrido en el que las células T senescentes liberan una tormenta de citocinas que pueden provocar la senescencia acelerada de otras células y tejidos98.
El TFG-β también desempeña un papel protrombótico correlacionado con el envejecimiento99. La estimulación de las células epiteliales del túbulo renal con TGF-β genera un incremento de la producción de ROS y de nicotinamida adenosina dinucleótido fosfato oxidasa (NADP oxidasa), lo que provoca el envejecimiento vascular y la reducción de la longitud telomérica, así como la activación de la ruta p53/2189,100. En condiciones normales, VEGF y TGF-β, junto con otros factores, como el factor de crecimiento epidérmico, participan en la curación de heridas al inducir la diferenciación de los miofibroblastos e incrementar los depósitos de colágeno101,102, pero, en fases avanzadas de este proceso, los fibroblastos senescentes junto con el TGF-β van a generar una fibrosis excesiva y una diferenciación epitelio-mesenquimal anormal que se correlaciona con la nefropatía. Además, el TGF-β producido por los podocitos combinado con las ROS de las células endoteliales glomerulares causan una segmentación de los capilares glomerulares, lo que genera un fallo renal y proteinuria masiva en el paciente103.
En pacientes renales tiene lugar lo que se denomina un fenotipo SIPS (stress-induced premature senescence), cuya diferencia con el fenotipo SASP radica en que SASP se produce por un envejecimiento natural, mientras que el SIPS es consecuencia de un daño o estrés, en este caso, principalmente por las toxinas urémicas99,104.
Vesículas extracelulares en la senescencia celularLa senescencia celular (desencadenada por replicación o por otros estímulos, como, por ejemplo, toxinas urémicas) aumenta de forma considerable la secreción de exosomas y MV en la mayoría de los casos. Como consecuencia, se altera la capacidad regenerativa de la vasculatura, especialmente la de las células endoteliales, que disminuyen su capacidad de migración celular y su potencial para formar estructuras vasculares104. Además, la cantidad de VE aparece modificada en múltiples enfermedades, incluida la ERC y, por ello, se está estudiando su posible papel como biomarcador predictor de enfermedades o de la progresión de la enfermedad. En concreto, en pacientes con ERC se ha descrito un aumento de la liberación de VE endoteliales que se asocia con disfunción endotelial producida por la uremia y la inflamación persistente de estos pacientes105.
No solo se ve alterado el número de VE en los enfermos renales, también su contenido, que genera procesos de disfunción endotelial, fibrosis y calcificación vascular106,107. Se ha descrito que uno de los desencadenantes de la calcificación vascular es la senescencia de las células de la vasculatura y que está asociado a cambios en la expresión de determinados miRNA, como es el caso del miRNA-146b-5p y miRNA-223-3p, y estos miRNA pueden transportarse en VE40.
Calcificación vascular en la enfermedad renal crónicaLa alteración vascular más prevalente en pacientes con ERC en estadios avanzados es la calcificación vascular, que en muchos casos no es detectada hasta que el daño es irreversible. La calcificación vascular se produce debido al aumento de proteínas morfogénicas de hueso y a la presencia de un alto contenido de calcio. El aumento en plasma de la ratio Ca/P produce un incremento de la expresión de proteínas que participan en la formación del hueso, como osteopontina, osteocalcina, ciertos proteoglicanos y proteína morfogénica ósea 2, al igual que otros factores que participan en la calcificación vascular12.
Existen 2tipos de calcificaciones: a) la calcificación de la túnica media, conocida también como esclerosis de Mönckeberg, que afecta a las CMLV y fibras elásticas, que se endurecen y pierden capacidad de distensión108 y b) la calcificación aterosclerótica de la túnica íntima, que se asocia con el depósito de lípidos y lipoproteínas seguido del depósito de calcio debajo de la túnica íntima109. Este depósito puede estimular el desarrollo de respuestas inmunes, tanto innatas como adaptativas, e inducir en las células endoteliales y CMLV la expresión de moléculas proinflamatorias, lo que estimula la infiltración de monocitos/macrófagos en los tejidos. Ambos tipos son muy comunes en la ERC avanzada y pueden coexistir en un mismo vaso.
El aumento de fósforo (hiperfosfatemia) y el aumento de calcio (hipercalcemia) son 2de los principales promotores asociados con el desarrollo de la calcificación vascular en la ERC14,108. En pacientes que reciben tratamientos con vitamina D junto a sales de calcio por vía oral y que tienen un balance positivo de calcio durante la sesión de diálisis se favorece el desarrollo de calcificaciones110. En modelos in vitro se ha demostrado que el incremento del fósforo favorece la transformación de CMLV en células osteogénicas, con producción de matriz extracelular y posterior mineralización. Por tanto, la hiperfosfatemia y la hipercalcemia no solo generan depósitos de forma pasiva, sino que además intervienen en un proceso altamente regulado por el que las CMLV se diferencian a osteoblastos111. En ocasiones, la disminución de la excreción renal de fosfatos provoca un aumento de su nivel en sangre que, junto con la deficiencia de inhibidores circulantes de la calcificación o la baja producción local de estos, favorece la calcificación a través de la activación del toll-like receptor 4 (TLR-4) que a su vez estimula la expresión de NF-κB en las CMLV y supone un aumento de la liberación de citocinas proinflamatorias14,112.
En estudios in vitro se ha demostrado que toxinas urémicas derivadas de la guanidina (dimetilarginina asimétrica), ácido guanidinobutírico, guanidina y ácido guanidino acético son capaces de estimular la osteoclastogénesis113. La uremia también favorece la secreción del factor de diferenciación de osteoblastos, Cbfa1, junto con un aumento de la expresión de proteínas osteogénicas107,114. El IS en las CMLV estimula la proliferación y favorece la producción de TGF-β (que participa en procesos de fibrosis)115. También, el incremento del estrés oxidativo (situación observada en los pacientes con ERC) está estrechamente asociado con el desarrollo de la calcificación vascular mediado por la expresión de Runx2 (runt-related transcription factor 2) en las CMLV108. Por tanto, diversos estímulos, como el aumento de ROS, las quimiocinas y citocinas proinflamatorias (CCL5, CCL2, TNF-α, IL-6) y los niveles de fosforo y calcio van a facilitar la entrada de NF-κB al núcleo de las CMLV, lo que estimula la producción de factores que participan en la calcificación (Runx2, OPN, Msh homrbox protein 2 o MSX2), fosfatasa alcalina, proteína morfogénica ósea 2) (fig. 4). Además, el calcio ayuda en la liberación de VE, lo que supone un refuerzo del proceso de calcificación107,115-117. Las propias CMLV liberan una mayor cantidad de VE en respuesta a un estrés por calcio ambiental, lo que también favorece la calcificación vascular118.
Por otra parte, la senescencia también desempeña un papel clave en el desarrollo de calcificación vascular. Se ha observado que las VE liberadas por macrófagos de personas envejecidas participan en la calcificación de la matriz vascular104. También las MV endoteliales encontradas en plasma de sujetos ancianos y liberadas por células endoteliales senescentes ayudan a la calcificación de las CMLV, ya que contienen altos niveles de calcio, grupos fosfato y proteínas, que participan en la iniciación de la calcificación12.
De manera que la alteración mineral, la uremia y el incremento del estrés oxidativo, la inflamación y la liberación de VE que presentan los pacientes con ERC favorecen la aparición de calcificación vascular y, con ello, se incrementa el riesgo de daño cardiovascular.
Vesículas extracelulares en la progresión de la enfermedad cardiovascular en pacientes con enfermedad renal crónicaDebido al incremento en la prevalencia de ERC es necesario identificar nuevas dianas terapéuticas que consigan frenar o revertir la progresión de la enfermedad119. Al igual que para estas enfermedades, las VE se han propuesto como biomarcadores de predicción y diagnóstico precoz de la ERC. Las VE promueven alteraciones en la señalización intercelular y el daño vascular, con el subsiguiente desarrollo de las ECV11,40. De este modo, ante una señalización intercelular anómala, se podrían identificar dianas terapéuticas que conducirán a un método de diagnóstico y una monitorización precoz para pacientes con ECV asociadas a la ERC40.
Las VE, como mensajeros intercelulares que son, pueden interaccionar con distintos tipos celulares a través de receptores de membrana o siendo internalizadas por la célula diana por distintos mecanismos (fusión de membranas, endocitosis, fagocitosis, micropinocitosis). Al final, el resultado de esta interacción entre VE y células es la activación o regulación de distintas rutas de señalización en la célula diana120.
Microvesículas endoteliales en el desarrollo de enfermedades cardiovasculares: disfunción endotelialLas toxinas urémicas, entre las que destacan el IS y el p-cresol, van a dañar a las células endoteliales, generando un estado proinflamatorio. Este daño continuo de las células endoteliales va a estimularlas para que liberen una mayor cantidad de MV que, a su vez, modifican el tono vascular y contribuyen a acrecentar el daño vascular, por lo que se acaba generando un círculo vicioso11,59,121-123. Estas MV endoteliales están implicadas en la etiopatogenia de varias ECV, entre ellas, la aterosclerosis11,124. En general, existe una relación entre la función renal y el riesgo cardiovascular, todo ello reflejado en una excesiva vesiculación endotelial que podría actuar como un marcador de disfunción endotelial.
Se ha descrito que las MV endoteliales promueven calcificación vascular, ya que el tratamiento de CMLV con MV endoteliales procedentes de células endoteliales previamente tratadas con IS indujo calcificación en las CMLV, a diferencia de los controles, en las que las MV endoteliales procedían de células endoteliales no tratadas con toxinas urémicas107.
Microvesículas plaquetarias en el desarrollo de enfermedades cardiovasculares: coagulaciónOtro tipo de MV implicadas en el desarrollo de ECV son las MV plaquetarias. En este proceso patológico se va a desencadenar una excesiva activación plaquetaria, que genera agregación plaquetaria, la secreción de sus gránulos internos y la liberación de un mayor número de MV, todo ello con actividad procoagulante120. Además, en una situación de disfunción endotelial, las plaquetas tienden a adherirse a la superficie dañada125 con la consiguiente liberación de potentes factores mitogénicos que conducen a la proliferación de las CMLV y a la progresión del daño vascular.
La propia técnica de diálisis puede constituir un factor de riesgo en los enfermos renales. Hay varios estudios que señalan la existencia de hipercoagulación en estos pacientes, probablemente debida a una mayor expresión del factor tisular126, que también es conocido como tromboplastina. Se expresa en la membrana celular y es sintetizado por diferentes tipos celulares, aunque también puede encontrarse en la superficie de monocitos y células endoteliales en estados inflamatorios. Este factor participa en la formación de trombos locales. Las VE expresan en su superficie factor tisular, por lo que pueden unirse a la superficie plaquetaria de un trombo en evolución, suceso muy común en pacientes urémicos127. Asimismo, se ha descrito una relación del factor tisular con pacientes urémicos; en concreto, se ha demostrado que altos niveles de este marcador, importante en la actividad trombogénica, pueden ser clave en aquellos pacientes en tratamiento renal sustitutivo según la técnica de diálisis recibida11. Esto indida que las VE que expresan factor tisular pueden ser biomarcadores de eventos trombóticos en pacientes urémicos127.
Vesículas extracelulares como biomarcador diagnósticoEn los últimos años, el análisis de las VE en los fluidos corporales se ha utilizado como una herramienta de diagnóstico (biomarcador) en diversas enfermedades. Es el caso de pacientes con ECV asociada a ERC40,128. Los biomarcadores son una herramienta analítica que informa de manera objetiva, medible y evaluable sobre la función biológica, algunos procesos patológicos o la respuesta a un tratamiento. Los biomarcadores son fundamentales en clínica, ya que permiten una identificación temprana de una enfermedad y, con ello, se consigue una intervención terapéutica precoz que evite el desarrollo de la enfermedad y sus posibles consecuencias sobre el individuo11. En general, se han detectado niveles altos de VE en numerosas enfermedades, destacando aquellas de carácter crónico e inflamatorio, como la diabetes, la hipertensión, el cáncer y la ERC129.
En el contexto de ERC se observa una relación entre la función renal y el riesgo cardiovascular, todo ello reflejado en una elevada producción de VE endoteliales que podría actuar como marcador de disfunción endotelial105. En concreto, Mezentsev et al. describieron que los niveles fisiológicos de MV endoteliales en los individuos sanos es de entre 103 y 104 MV/mL, mientras que en los individuos con ECV es de 105 MV/mL130. Diferentes estudios señalan la existencia de una mayor concentración de VE circulantes en los pacientes con ECV, como la insuficiencia cardiaca congestiva, enfermedad arterial coronaria, enfermedad vascular periférica e isquemia cerebral60.
En pacientes urémicos existen también altos niveles de MV plaquetarias con efecto procoagulante, lo que puede estar asociado a trastornos de la coagulación, accidentes cerebrovasculares, angina inestable e infarto agudo de miocardio. Esto refuerza su posible contribución al desarrollo de la ECV (sobre todo asociado a trombosis y desestabilización plaquetaria) y a su uso como posible biomarcador diagnóstico131,132. En particular, los niveles de MV plaquetarias en pacientes en HD y diálisis peritoneal continua ambulatoria eran mayores que en las personas sanas y aún mayores en aquellos pacientes con un evento trombótico131. También se ha observado que pacientes con ERC e infarto agudo de miocardio y, por ello, con un riesgo elevado de nuevo evento trombótic, presentan niveles elevados de MV plaquetarias y que los niveles de MV plaquetarias son más elevados en aquellos pacientes con una ERC más avanzada133. Estas MV plaquetarias expresan P-selectina, un factor que indica activación plaquetaria. Además, como ya se ha mencionado, el aumento de VE con factor tisular puede ser un marcador clínico de riesgo de accidente trombótico en pacientes con ERC127.
Terapias basadas en vesículas extracelularesSe ha propuesto que las VE y su contenido pueden actuar como dianas terapéuticas. En primer lugar y puesto que se ha relacionado la aparición de ECV con el aumento de la liberación de VE, se pueden diseñar estrategias farmacológicas que inhiban su liberación, o incluso, su recepción por las células diana. Por ejemplo, se puede inhibir la ceramida, un componente importante en la biogénesis de los exosomas63. Aun así, los tratamientos enfocados en la liberación de VE son escasos, debido a la falta de conocimiento concreto de los mecanismos que intervienen en la biogénesis y la liberación de las VE11. Tratamientos enfocados en la reducción de la liberación de VE podrían disminuir la morbimortalidad de los pacientes con ERC63.
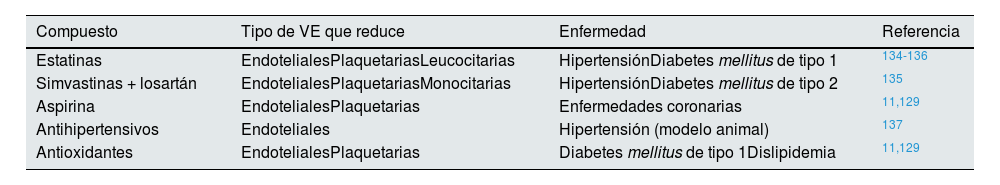
Se ha observado que algunos fármacos que se han empleado de manera tradicional para tratar ECV y la ERC tienen la capacidad de modular la liberación de VE (tabla 3).
Tratamientos que reducen la liberación de vesículas extracelulares a sangre
| Compuesto | Tipo de VE que reduce | Enfermedad | Referencia |
|---|---|---|---|
| Estatinas | EndotelialesPlaquetariasLeucocitarias | HipertensiónDiabetes mellitus de tipo 1 | 134-136 |
| Simvastinas + losartán | EndotelialesPlaquetariasMonocitarias | HipertensiónDiabetes mellitus de tipo 2 | 135 |
| Aspirina | EndotelialesPlaquetarias | Enfermedades coronarias | 11,129 |
| Antihipertensivos | Endoteliales | Hipertensión (modelo animal) | 137 |
| Antioxidantes | EndotelialesPlaquetarias | Diabetes mellitus de tipo 1Dislipidemia | 11,129 |
Se han realizado estudios para verificar si los niveles de MV endoteliales podrían ser modulados mediante algún fármaco. Por ejemplo, el uso de estatinas, concretamente la atorvastatina, demostró una disminución en los niveles de VE endoteliales y VEGF134. Este fenómeno se produce gracias a que las estatinas inhiben la vía de las Rho-cinasas, implicadas en la reorganización del esqueleto y en la liberación de las MV y, por tanto, disminuyen la liberación de MV a sangre (ya no solo las endoteliales, también las derivadas de plaquetas y leucocitos)135,136. Además, en pacientes con hipertensión y diabetes mellitus de tipo 1, se ha observado que el tratamiento con simvastatina junto con losartán disminuye las concentraciones en plasma de VE endoteliales, plaquetarias y derivadas de monocitos135. La aspirina también podría tener un efecto reductor de la liberación de VE, en concreto, las endoteliales y plaquetarias, pero los resultados son algo heterogéneos11,129. También se observó una reducción de MV endoteliales en sangre periférica en un modelo animal de hipertensión en el que se usaron fármacos antihipertensivos (aliskiren, nerbivolol y olmesartan); esta reducción también mediaba los efectos antiangiogénicos de estos fármacos137.
Los antioxidantes, como la vitamina C, también podrían tener un efecto beneficioso en este sentido, disminuyendo la liberación de VE; en concreto, se ha observado una disminución de VE endoteliales y plaquetarias en pacientes con diabetes mellitus de tipo 1 y con dislipidemia tras sufrir un infarto de miocardio11.
Además de usar las VE como diana de los tratamientos, recientemente se ha estado investigando el uso de las VE como una herramienta terapéutica, como una forma de administrar un tratamiento de forma más efectiva y «fisiológica». Esta idea surge de que las VE en condiciones fisiológicas pueden tener efectos beneficiosos sobre las células diana. Por ejemplo, se ha observado que las VE pueden participar en procesos de reparación tisular y modulación inmunológica, ya que podrían utilizarse directamente como agentes terapéuticos en la medicina regenerativa y el tratamiento de enfermedades autoinmunes63.
En condiciones fisiológicas, las MV derivadas de plaquetas son capaces (a través del VEGF) de estimular la proliferación, supervivencia, migración de células y formación de nuevos capilares. En cambio, las MV endoteliales se han relacionado con procesos regenerativos y con el mantenimiento de la homeostasis vascular. Por tanto, un aumento en el nivel de este tipo vesicular es clave en procesos en los que se está llevando a cabo una regeneración vascular11,120.
En el riñón, en modelos de nefropatía diabética, ERC, fibrosis y de daño renal agudo, se ha observado que las VE derivadas de células madre mesenquimales presentan una función renoprotectora. En general, las VE derivadas de células madre son las que presentan unos mayores efectos beneficiosos en enfermedades renales. Esto se puede deber a su contenido rico en miRNA y proteínas (principalmente relacionadas con proliferación celular, migración y adhesión)63.
Por último, también se ha planteado el uso de las VE como vehículo de tratamientos (que pueden ser fármacos, miRNA o proteínas). Este tipo de administración tiene como ventaja que son menos inmunogénicas y citotóxicas, y hasta la fecha parecen tener menor rechazo que las nanopartículas63.
ConclusionesLos pacientes con ERC, debido a la uremia, presentan un aumento de la inflamación y del estrés oxidativo y, en consecuencia, un envejecimiento prematuro, con acumulación de células senescentes, que explica la aparición precoz de enfermedades asociadas a edades más avanzadas, como, por ejemplo, la ECV. El antecedente de las ECV, tan prevalentes en enfermos renales, es el daño endotelial producido a consecuencia del estrés oxidativo, la inflamación crónica y las VE liberadas debido al daño celular. Estas VE contienen miRNA, como es el caso del miR-21 y el miR-155, que alteran la expresión de distintas proteínas y que pueden ser responsables del aumento de la permeabilidad vascular y del mantenimiento de la inflamación.
Los pacientes con ERC presentan un mayor número de VE en plasma; en concreto, destacan las MV endoteliales, que aumentan debido al daño endotelial de estos pacientes y suponen un primer paso hacia complicaciones vasculares más graves, como la calcificación vascular. Las MV endoteliales de pacientes con ERC tienen unos altos contenidos de calcio y proteínas de unión al calcio que facilitan o promueven la calcificación vascular y, en consecuencia, pueden desencadenar ECV. En estos pacientes también existen alteraciones en la coagulación, consecuencia de la disfunción endotelial y de las MV plaquetarias, que aparecen en mayor cantidad en el plasma de pacientes urémicos. Las MV plaquetarias también podrían participar en la formación de placas de ateroma y en la calcificación vascular.
Con estos antecedentes, la medición de MV puede ser un buen predictor de complicaciones cardiovasculares en pacientes con ERC, al posibilitar su detección temprana y facilitar un abordaje terapéutico precoz. Aun así, quedan por fijar umbrales a partir de los cuales predecir si existe mayor riesgo de sufrir ECV para que sea posible el traslado de estos pacientes al ámbito clínico. Las VE también podrían ser una buena diana terapéutica para frenar de forma temprana el desarrollo de ECV, pero para ello se requiere un mayor avance en la investigación de fármacos que modulen la liberación de VE. Aun así, ya se conocen fármacos comúnmente desarrollados para tratar ECV o sus factores de riesgo que modulan la liberación de VE y que, por tanto, tienen un efecto beneficioso en estos pacientes tratados. También se ha demostrado que las VE liberadas principalmente por células madre tienen un contenido beneficioso para el individuo, por lo que podrían ser interesantes para tratar a pacientes con ERC que están altamente predispuestos a sufrir ECV. Incluso se ha planteado el uso de las VE como vehículo que permita dirigir los fármacos de un determinado tratamiento de forma más específica.
En resumen, el uso de VE como marcadores para la predicción, diagnóstico, pronóstico y monitorización de terapias de enfermedades complejas resulta cada vez más atractivo, así como su potencial para identificar nuevas dianas terapéuticas. En este sentido, la evidencia clínica y experimental señala que las VE pueden ser de utilidad en el control de la eficacia de las terapias sustitutivas; de hecho, los resultados obtenidos hasta la fecha en los diversos estudios son muy prometedores.
FinanciaciónEste trabajo ha sido financiado por el Instituto de Salud Carlos III a través del proyecto PI17/01029, PI19/00240 y PI20/01321 (cofinanciado por la Unión Europea, FEDER/FSE «Una manera de hacer Europa»/ «El FSE invierte en tu futuro») y la Sociedad Española de Nefrología (Proyecto Fundación Senefro).
Programa RICORS (RICORS2040), fondos FEDER, Instituto de Salud Carlos III (ISCIII) (RD21/0005/0002).
AF es una becaria del programa Contratos Predoctorales de Investigación en Salud del Instituto de Salud Carlos III (FI20/00018). GV recibió una beca de la Comunidad de Madrid y el Fondo Social Europeo (PEJ-2020-AI/BMD-18141). NS tiene una beca del programa Becas Asociadas a Proyectos de Investigación del Instituto de Investigación Sanitaria Hospital 12 de Octubre (i+12) (Madrid, España).
Conflicto de interesesLos autores no declaran conflicto de intereses.



