




Describimos el caso de una mujer joven, que fue diagnosticada de insuficiencia renal avanzada, con un hallazgo casual de una nefrocalcinosis sin una etiología clara, al haberse encontrado asintomática a lo largo de su vida. El estudio genético por paneles de genes conocidos asociados a enfermedad tubulointersticial permitió descubrir una acidosis tubular renal distal autosómica dominante, asociada a una mutación de novo en el exón 14 del gen SLC4A1, que hubiera sido imposible diagnosticar clínicamente por lo avanzado de la enfermedad renal cuando fue descubierta.
We describe the case of a young woman who was diagnosed with advanced kidney disease, with an incidental finding of nephrocalcinosis of unknown aetiology, having been found asymptomatic throughout her life. The genetic study by panels of known genes associated with tubulointerstitial disease allowed us to discover autosomal dominant distal renal tubular acidosis associated with a de novo mutation in exon 14 of the SLC4A1 gene, which would have been impossible to diagnose clinically due to the advanced nature of the kidney disease when it was discovered.
La nefrocalcinosis —incremento generalizado de contenido de calcio en el riñón— es la consecuencia de diversos factores que contribuyen al daño renal (nefrolitiasis: incremento en la concentración urinaria de calcio/oxalato/fosfato y, sobre todo, por graves defectos metabólicos)1. Entre las principales causas hereditarias de nefrocalcinosis se incluyen la deficiencia de adenina fosforibosiltransferasa, cistinuria, enfermedad de Dent, hipomagnesemia-hipercalciuria-nefrocalcinosis familiar, hiperoxaliuria primaria y acidosis tubulares renales (ATR)2,3.
Dentro de estas últimas causas, la ATR distal primaria o hereditaria ha cobrado especial interés en los últimos años por los avances en el conocimiento de los mecanismos moleculares, que han permitido conocer mutaciones de las principales proteínas implicadas en el trasporte ácido-base. La ATR se manifiesta en ambas formas hereditarias autosómica dominante (AD) y recesiva (AR), asociadas a mutaciones en los genes SLC4A1, ATP6V1B1, CA2 y ATP6V0A4. Se caracteriza por una acidosis metabólica hiperclorémica, debida a un fallo en la eliminación urinaria de hidrogeniones [H+]). Los principales síntomas que acompañan a la ATR distal son retraso del crecimiento, vómitos/diarrea o estreñimiento, falta de apetito, poliuria y polidipsia, y nefrocalcinosis. El pronóstico de la ATR distal es bueno si se diagnostica y trata con bicarbonato/citrato potásico a edades tempranas4. Sin embargo, en pacientes asintomáticos a lo largo de los años puede resultar difícil establecer mediante estudios clínicos/analíticos la causa primaria una vez que manifiestan insuficiencia renal avanzada, que se asocia a un pronóstico no tan benigno. Una forma útil de establecer el diagnóstico en este tipo de situaciones es por medio del diagnóstico genético que englobe la totalidad de los genes conocidos asociados a dicha enfermedad.
En este artículo describimos el caso de una mujer joven, sin antecedentes familiares de enfermedad renal, sin estudio nefrológico previo, asintomática a lo largo de su vida, en la que al descubrir una insuficiencia renal avanzada se comprobó que la paciente tenía calcificado el parénquima renal. La realización de un estudio genético de todos los genes conocidos asociados a nefrocalcinosis diagnosticó a la paciente como un caso de ATR-AD, identificando una mutación de novo en el gen SLC4A1como causa primaria de su nefrocalcinosis.
Caso clínicoAntecedentesMujer de 26 años, sin antecedentes relevantes, ni tratamiento crónico, que consultó en Atención Primaria por malestar general, calambres y parestesias en manos y pies y, desde hacía 2días, también refería imposibilidad para elevar el párpado izquierdo. En Atención Primaria se solicitó un control analítico y se detectó una creatinina de 4,6mg/dl (el único control previo registrado del que disponíamos en nuestro centro era de 6 años antes con una creatinina de 1,5mg/dl), motivo por el cual se derivó a la paciente a Urgencias. No se constataron antecedentes familiares de enfermedad renal en ningún miembro de la familia.
Evolución clínicaSu presión arterial fue de 135/67mmHg, frecuencia cardíaca 120 lpm y temperatura de 37,5°C, con una exploración física anodina. La analítica en sangre repetida en Urgencias confirmó el deterioro de la función renal (creatinina 5,1mg/dl y urea 245mg/dl), junto con sodio de 124mmol/l; potasio de 3,5mmol/l; pH de 7,08; bicarbonato de 5,9mmol/l; pCO2 de 20mmHg; calcio iónico corregido de 0,69mmol/l (normal 1,13-1,32); magnesio de 1mg/dl; leucocitos de 14.060 y el resto de hemograma y el estudio de coagulación normales. Otras determinaciones analíticas diferidas mostraban: calcio 7,4mg/dl; fósforo 5,6mg/dl; PTHi 298pg/ml; vitamina D 18,27ng/ml y ácido úrico 8,8mg/dl. El estudio inmunológico (inmunoglobulinas, ANA) era normal. En la electroforesis en sangre: disminución de gammaglobulinas. La serología de virus B, C y VIH fue negativa. En el sistemático de orina tenía pH 6,0, sangre+, leucocitos +++. En la orina de 24 h, la proteinuria era de 0,73g, el aclaramiento de creatinina de 12ml/min, la uricosuria de 600mg, la calciuria<4mg/kg y la oxaluria 10mg (normal: 4-31).
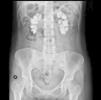
El electrocardiograma mostraba ritmo sinusal, con elevación de ST de 1mm en todas las derivaciones. La radiografía de tórax fue normal. En la radiografía simple de abdomen se comprobaron extensas calcificaciones renales bilaterales (fig. 1). Tras corrección de hipocalcemia, hipomagnesemia y acidosis metabólica la paciente fue dada de alta, sin incidencias relevantes durante el ingreso. Los valores analíticos al alta fueron creatinina 3,6mg/dl; sodio 142mmol/l; potasio 4,4mmol/l; pH 7,43; bicarbonato 24,6mmol/l; calcio 8,6mg/dl; calcio iónico corregido 1,16mmol/l; fósforo 2,9, PTHi 103ng/ml y magnesio 1,9mg/dl. El tratamiento al alta fue: carbonato cálcico 2,5 g cada 8 h; calcitriol 0,5 mcg/día; bicarbonato sódico 1 g/día y suplementación de potasio y magnesio oral.
Al cabo de unas 48 h, después de haber sido dada de alta del Servicio de Nefrología, consultó en el Servicio de Urgencias por imposibilidad tanto para hablar como para realizar movimientos linguales. Además, la paciente refería que la noche anterior presentaba alteración en el control de movimientos de la mano derecha. Asimismo, asociaba dificultad para la deglución y astenia intensa. No presentaba fiebre ni cefalea. En la exploración física la presión arterial fue de 102/72mmHg, frecuencia cardiaca 75 lpm, saturación de oxígeno basal 98% y el resto de la exploración, normal. La analítica en sangre fue: creatinina 3,6mg/dl; sodio 141mmol/l; potasio 4,4mmol/l; pH 7,43; bicarbonato 24,6mmol/l; calcio 8,6mg/dl; calcio iónico corregido 1,16mmol/l y magnesio 1,9mg/dl. La paciente fue valorada por el Servicio de Neurología y quedó ingresada a su cargo para estudio. En dicho servicio se realizó test de edrofonio (negativo), TAC cerebral sin contraste (sin hallazgos significativos), EEG (abundantes brotes de actividad paroxística [ondas theta] en región temporal izquierda, con propagación al resto del hemisferio, así como a región homóloga contralateral, formando algunos brotes paroxísticos bilaterales de ondas agudas). Se inició tratamiento con levetiracetam 250mg/12 h, que resultó insuficiente para el control de las crisis. La paciente evolucionó a status focal, sin respuesta a diacepam (hasta 10mg) ni a bolos de ácido valproico, por lo que precisó traslado a la UCI. La actividad comicial consiguió controlarse tras el inicio de perfusión de ácido valproico (1g/24 h), desde entonces la paciente permanece asintomática. Posteriormente, en planta de Neurología se controló con tratamiento oral: permaneció asintomática y pudo ser dada de alta con 500mg/12 h por vía oral de ácido valproico.
Estudio genéticoDada la ausencia de antecedentes familiares de enfermedad renal, la situación actual de insuficiencia renal avanzada y, por tanto, las dificultades diagnósticas para hacer estudios específicos que permitieran aclarar cuál fue el proceso primario que había propiciado el desarrollo de nefrocalcinosis, se decidió realizar un estudio genético. Se solicita el análisis de todos los genes asociados a enfermedad tubulointersticial conocidos (http://nefrochus.villaweb.es/en/) que incluye el análisis de las variantes génicas encontradas en los genes conocidos asociados a las formas de nefrocalcinosis (Nefrochus, Santiago de Compostela). La realización del estudio determinó que la paciente (II:1) era portadora de una mutación missense en el exón 14 del gen SLC4A1, asociada a acidosis tubular renal distal autosómica dominante (ATR-AD). Dicha mutación (c.1766G>A, fig. 2A) supone la sustitución de un aminoácido básico (p.R589H) arginina (Arg, R) por un aminoácido histidina (His, H) no encontrado previamente en cromosomas normales de nuestra cohorte, ni en el proyecto internacional 1000 genomes, ni en la base de datos dbSNP137, pero sí descrito en la literatura5,6, con un código genómico rs121912744 catalogado como CLINSIG=pathogenic y asociado a familias con acidosis tubular renal distal autosómica dominante. El estudio funcional realizado con diversas herramientas de predicción in silico adjudica a la variante génica criterios de patogenicidad (SIFT: deleterious; polyphen2: deleterious; LRT: deleterious; MutationTaster: disease_causing_automatic; mutation assessor: medium; FATHMM: tolerated; RadialSVM: deleterious y LR: deleterious).
Estudio de cosegregación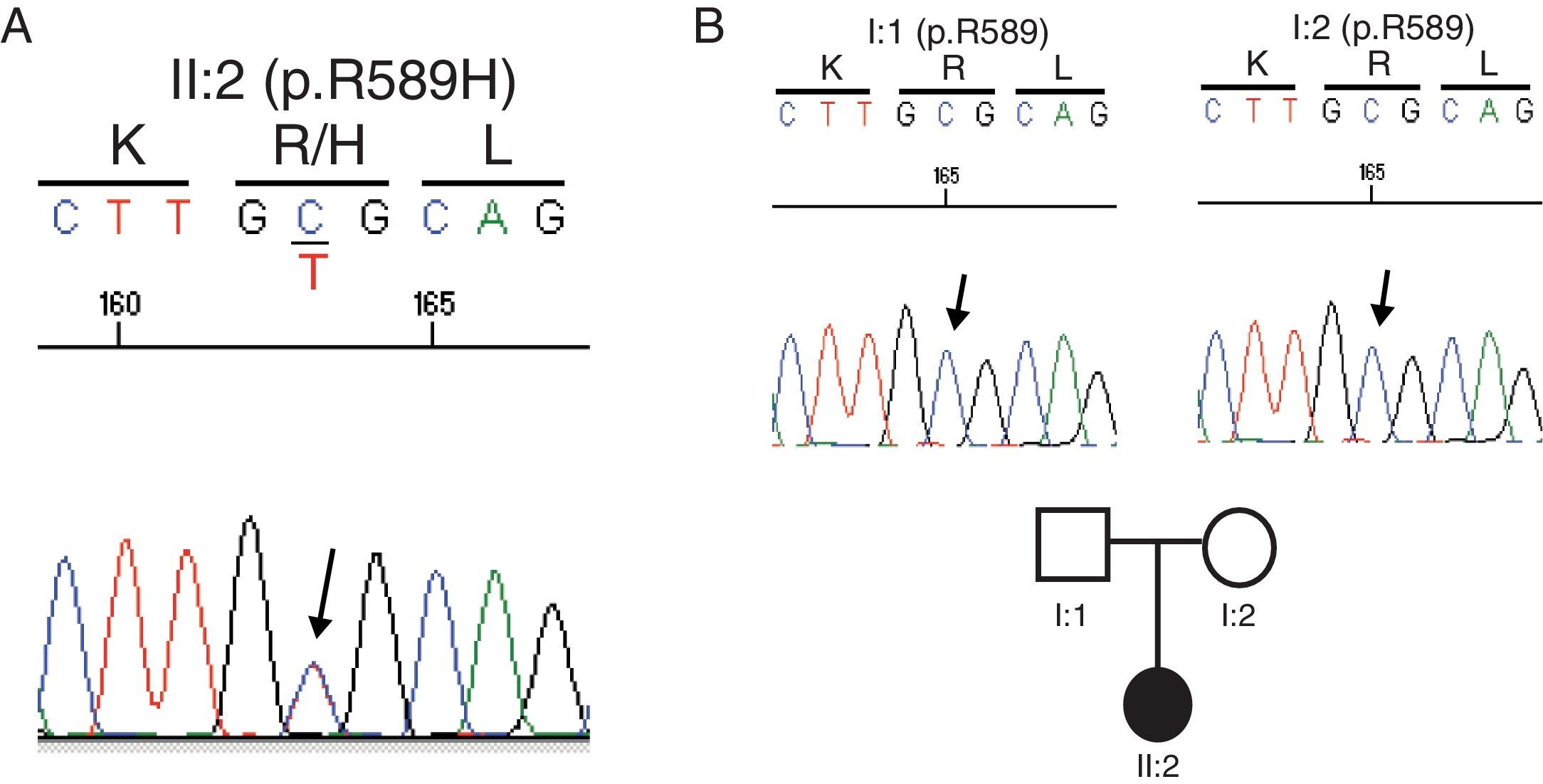
De cara a poder decidir sobre una posible donación de trasplante renal de vivo, se realiza un estudio de cosegregación en ambos progenitores (I:1 y I:2). El no ser ninguno portador de la mutación R589H indica que dicha mutación es de novo o espontánea, y así pueden ser candidatos a donación (fig. 2B).
DiscusiónEl estudio genético a través del uso de un panel para todos los genes conocidos hasta el momento asociados a nefropatía tubulointersticial nos ha permitido conocer que la causa primaria de la nefrocalcinosis asociada a nuestra paciente se debe a una acidosis tubular renal distal por una mutación missense de novo en el gen SLC4A1 previamente descrita en la literatura5,6.
En la clínica, la ATR distal puede diagnosticarse determinando la creatinina plasmática y las excreciones fraccionales de sodio, potasio, cloro, la calciuria y la citraturia y, para los casos de dudas, se pueden realizar pruebas de acidificación tubular con administración de NH4Cl. Sin embargo, en pacientes asintomáticos, y sin antecedentes familiares, esta entidad puede pasar desapercibida y una vez que se llega a una situación de insuficiencia renal avanzada, las pruebas funcionales pueden estar artefactadas o no ser útiles para llegar al diagnóstico exacto que ha provocado la nefrocalcinosis, como el caso que presentamos, al tener una acidosis metabólica multifactorial y tubulopatía por el daño renal establecido. De hecho, en nuestra paciente la aparición posterior de crisis convulsivas nos llevó a considerar la hipomagnesemia-hipercalciuria-nefrocalcinosis familiar como posible causa de su nefrocalcinosis, que finalmente se excluyó al hacer el estudio genético.
Este trabajo pone de manifiesto la importancia de hacer estudios genéticos, ya que nos ha permitido conocer la causa primaria asociada a la que ha provocado la nefrocalcinosis. Así pues, reconocemos que la realización de test diagnósticos supone las siguientes ventajas: 1) en caso de haber podido realizarse el diagnóstico precozmente, podría haberse evitado que nuestra paciente llegara a una situación de insuficiencia renal tan avanzada, al haber podido instaurar tratamiento precoz. 2) En el caso de plantearse una terapia sustitutiva renal, podríamos estar más seguros de que la enfermedad no recidivaría en el injerto renal si conocemos la causa exacta que provocó la nefrocalcinosis. 3) También podríamos preseleccionar progenitores con total seguridad como posibles donantes, excluyéndolos en el caso de que estos fueran portadores de una mutación. En conclusión, describimos el caso de una paciente portadora de una mutación en el gen SLC4A1 asociada a acidosis tubular renal distal autosómica dominante, que provocó una nefrocalcinosis, y que había pasado desapercibida clínicamente hasta llegar a una fase de insuficiencia renal avanzada. Sin el estudio genético que incluya todos los genes asociados a enfermedad tubulointersticial, habría sido imposible llegar al diagnóstico certero de la causa primaria de nefrocalcinosis.
Conflicto de interesesNo existe conflicto de intereses ni financiación.



