






El caso fue presentado en la Web de Nefrología (www.revistanefrologia.com), y durante 15 días se realizaron distintos comentarios, todos ellos de gran interés, a los que puede accederse en la Web de Nefrología, en el apartado correspondiente al número 2, volumen 8, 2016 de NefroPlus.
CASO CLÍNICO
Paciente mujer de 24 años de edad, de profesión militar, sin antecedentes personales de interés excepto un primer embarazo sin complicaciones, que finalizó con un parto normal 15 meses antes de su ingreso. Su madre, de 55 años, fue diagnosticada a los 52 de enfermedad de Churg-Strauss sin afectación renal y su abuela materna falleció en hemodiálisis crónica por enfermedad renal terminal no filiada. No fuma ni consume alcohol o drogas. Desde hace 11 meses recibe anticonceptivos orales.
Siete días antes de su ingreso, después de un ejercicio físico intenso, presenta astenia, anorexia, náuseas y, 3 días más tarde, diarrea sin fiebre con cuatro deposiciones al día, líquidas y sin sangre, de 48 h de duración. A pesar del cese de la diarrea, su estado general no mejora, por lo que acude a su hospital de referencia donde se le objetiva oliguria e insuficiencia renal grave, hipertensión arterial, anemia y plaquetopenia. Fue tratada con fluidoterapia intravenosa (i.v.), trasfundida con una unidad de concentrado de hematíes y remitida a nuestro servicio.
Al ingreso, la paciente estaba consciente y orientada, pálida, sin disnea y toleraba el decúbito. La presión arterial fue de 170/110 mmHg, la temperatura de 36,5 ºC, la frecuencia respiratoria de 12 resp/min y la frecuencia del pulso de 70 lat/min. La exploración neurológica fue normal y el fondo de ojo no mostró alteraciones. La auscultación cardiopulmonar y la exploración abdominal fueron normales. Había edemas pretibiales ligeros. No se observaron adenopatías ni tampoco alteraciones en la piel ni en los pulsos periféricos.
Los datos analíticos iniciales más relevantes para el caso se recogen en la tabla 1. Posteriormente se comprobó que los ANA (anticuerpos antinucleares), ANCA (anticuerpos anticitoplasma de neutrófilos) y la serología para VHB (virus de la hepatitis B), VHC (virus de la hepatitis C) y VIH (virus de la inmunodeficiencia humana) eran negativos. El urocultivo fue negativo y el coprocultivo en medio enriquecido con sorbitol, tomado 5 días después del cese de la diarrea, mostró flora saprofítica normal. El electrocardiograma mostró un ritmo sinusal sin alteraciones. La ecografía renal evidenció unos riñones con ecoestructura y tamaño normales (12,3 cm el derecho y 12,5 cm el izquierdo). El resto de estudios de imagen (radiografía de tórax, ecografía abdominal y tomografía computarizada cerebral) fueron normales.
Fue tratada con plasmaféresis e infusión de plasma diarias, hemodiálisis en días alternos, amlodipino (10 mg/día), enalapril (40 mg/día) y doxazosina (8 mg/día). Después de 7 días de tratamiento, la situación de la paciente no mejoró. Mantuvo la plaquetopenia y necesitó de la administración de cinco unidades de concentrado de hematíes para controlar la anemia, continuó precisando diálisis y permaneció hipertensa. La tolerancia a la plasmaterapia no fue buena. En todas las sesiones se produjeron reacciones adversas en forma de exantema cutáneo y taquipnea, a pesar de la premedicación con esteroides y antihistamínicos. Finalmente, fue preciso suspender la sexta sesión por aparición de exantema cutáneo y broncoespasmo con la infusión de una pequeña cantidad de plasma. Por todo ello, al séptimo día del ingreso se decidió suspender la plasmaterapia, realizar un procedimiento diagnóstico e iniciar un nuevo tratamiento.
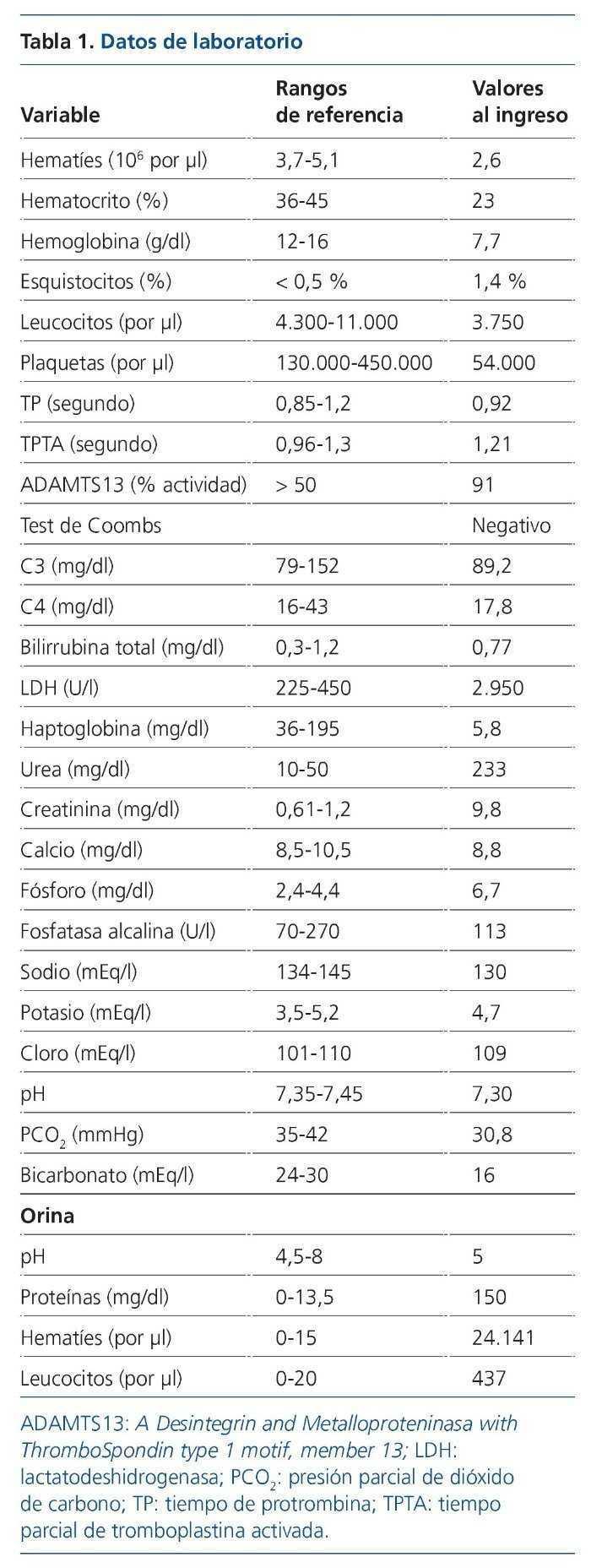
El procedimiento diagnóstico fue una biopsia renal que mostró hallazgos característicos de microangiopatía trombótica —MAT— (figura 1). El nuevo tratamiento con eculizumab 900 mg i.v. los días 7, 11, 16 y 23 del ingreso normalizó la lactatodeshidrogenasa (LDH) y la haptoglobina con la primera dosis, y las plaquetas y la hemoglobina después de la segunda (figura 2). La presión arterial se controló con la segunda dosis y la medicación antihipertensiva se suspendió al alta tras 24 días de ingreso. La función renal mejoró progresivamente, se pudo suspender la diálisis a partir de la segunda dosis y la creatinina plasmática y el aclaramiento de creatinina se normalizaron después de recibir otras seis dosis bisemanales de 1.200 mg i.v. de eculizumab en régimen ambulatorio (figura 3).
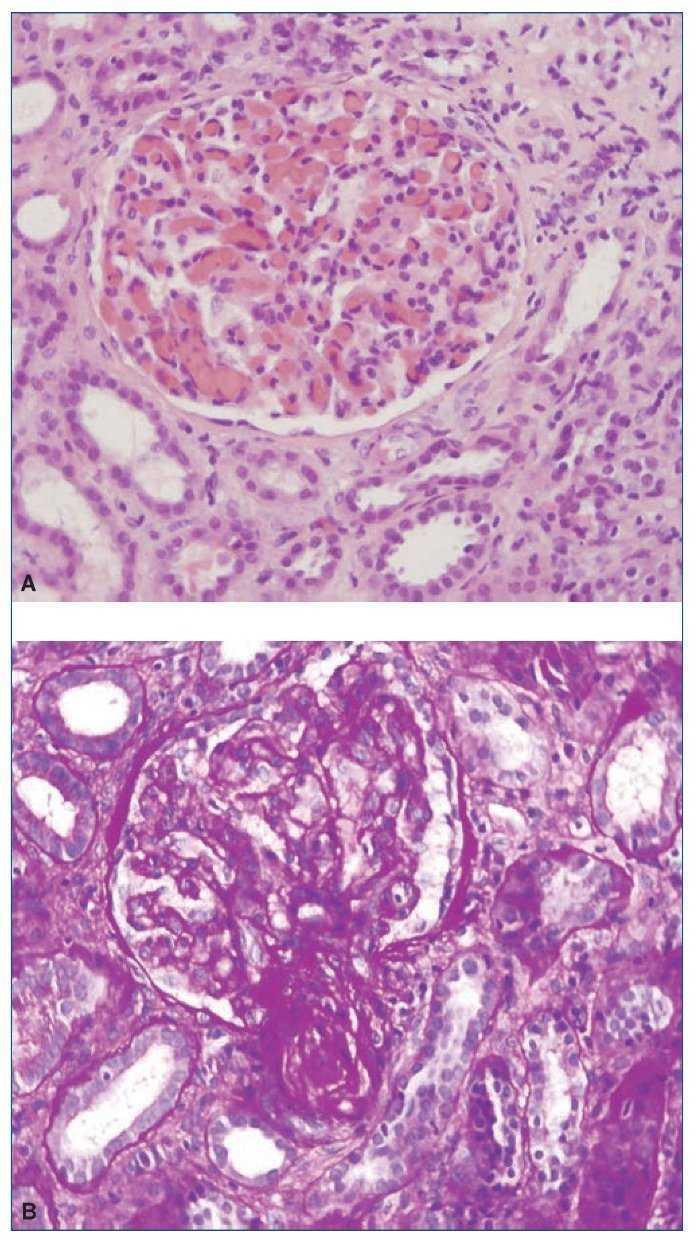
Figura 1. Biopsia renal.
A) Hematoxilina-eosina. Glomérulo con incremento moderado de la celularidad y marcada congestión capilar. B) PAS. Ensanchamiento mesangial con flexuosidad isquémica y reduplicación de membranas basales. La arteriola muestra una marcada reducción de la luz, engrosamiento subintimal y trombosis.
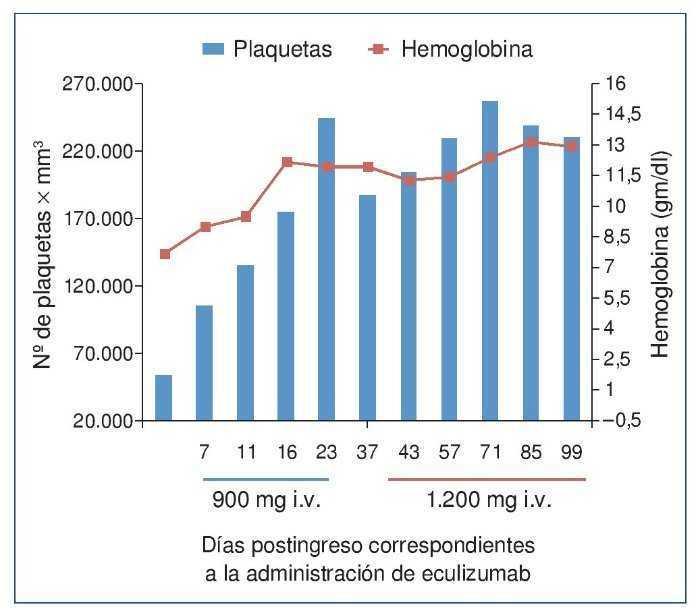
Figura 2. Evolución del recuento plaquetario y de la concentración de hemoglobina después de la administración de eculizumab.
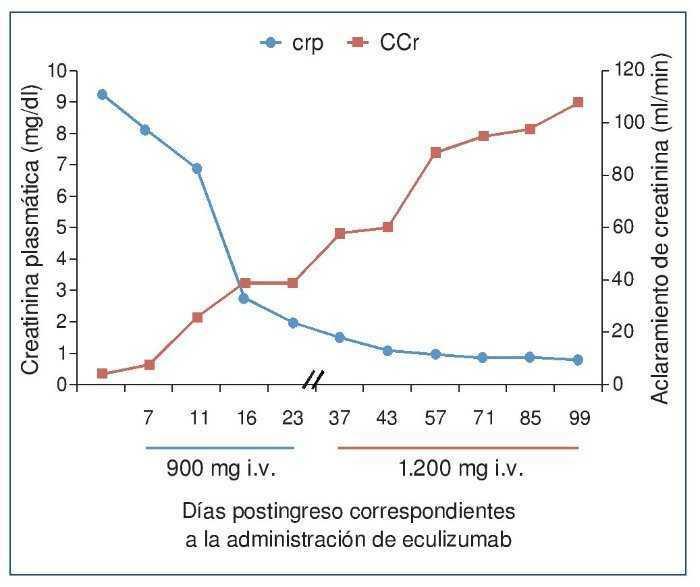
Figura 3. Evolución de la creatinina plasmática y del aclaramiento de creatinina después de la administración de eculizumab.
El estudio genético y funcional de la paciente reveló una mutación (c.286+2T>G) en el gen de MCP (gen de MCP cofactor proteína, CD46), con disminución del 55 % de la expresión de la proteína en la superficie de los leucocitos, un cambio previamente descrito de significado incierto (c.1534+5G>T) en CFI y un haplotipo de riesgo en heterocigosis en CFH (tabla 2). El estudio en su madre mostró los mismos cambios en MCP y CFI que en la paciente.
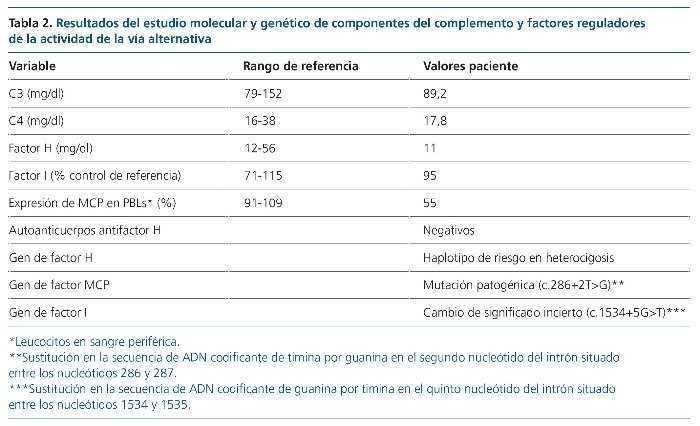
El diagnóstico definitivo fue de síndrome hemolítico urémico atípico (SHUa) mediado por el complemento, con componente familiar y asociado al uso de anticonceptivos orales.
DISCUSIÓN
Definición
El SHUa mediado por el complemento se caracteriza por anemia hemolítica microangiopática (AHM), trombocitopenia y daño renal agudo, que aparecen, cuando hay un factor desencadenante, en sujetos con alteraciones genéticas o adquiridas en el sistema del complemento1-4. Estos trastornos permiten la actividad incontrolada de C3 convertasa (C3bBb) de la vía alternativa del complemento en la superficie endotelial y la generación de C5a y C5-9b, que al inducir inflamación, daño en la membrana celular y activación de la vía de la coagulación causan una MAT4,5.
Patogenia
El 60 % de los pacientes con SHUa son portadores de una o más anomalías genéticas o adquiridas del complemento1,3,6-8. Las mutaciones genéticas se traducen en pérdida de función de proteínas plasmáticas (CFH, CFI)9,10 o de membrana (MCP, THMD)11,12, que interactúan para disociar a C3bBb o degradar a C3b en la superficie celular, o en ganancia de función de proteínas constituyentes de C3bBb (C3, CFB)13,14.
Las mutaciones más frecuentes son las que ocurren en la región C terminal de CFH (20-30 %), las de MCP con disminución de función o de expresión de proteína (10-15 %) y las de CFI (4-10 %), mientras que las mutaciones de C3 (2-10 %), THBD (3-4 %) y CFB (1-4 %) son más raras7,15. Algunos pacientes con SHUa son portadores de genes híbridos entre el de CFH y los de proteínas relacionadas con el complemento (CFHR1-5)16,17. Los anticuerpos anti-CFH son más frecuente en niños (25-50 %) que en adultos (5-10 %)18-20 y pueden asociarse a deleciones homocigóticas en el gen de CFHR1, CFHR-3 y CFHR4, aunque también se han descrito con mutaciones en CFH, CFI, MCP, CFB o C321-23.
La penetrancia, que se estima del 50 % entre sujetos portadores de anomalías del complemento, se incrementa si coexisten más de una mutación, si se combinan mutaciones y autoanticuerpos o en presencia de algunos polimorfismos de un único nucleótido o de haplotipos completos de CFH y MCP6.
El complemento podría estar implicado en algunos casos de SHU por Escherichia coli productora de toxina shiga (STEC), porque se ha observado que la toxina puede fijarse a CFH, interfiriendo con su función reguladora24. Se desconoce si el complemento puede jugar un papel en el SHUa asociado a mutaciones en diaglicerol cinasa (DGKE)25,26. Estos pacientes presentan daño endotelial con expresión de ICAM-1 y factor tisular mediado por p36 MAPK27, pero se han encontrado casos en los que coexisten mutaciones en DGKE y en C3 o trombomodulina28 y, recientemente, se ha descrito en una familia consanguínea una mutación en DGKE asociada al consumo de C329. Tampoco se conoce el alcance que puede tener el hallazgo de un estudio en el que la mitad de los pacientes con SHUa compartía mutaciones en el complemento y en genes del sistema de coagulación, principalmente de plasminógeno30.
Manifestaciones clínicas y evolución
El SHUa es una enfermedad muy rara. En Europa se estima una incidencia y una prevalencia de 0,11 y de 3,3 por millón de habitantes, respectivamente31. Afecta preferentemente a niños y adultos jóvenes, aunque puede manifestarse en cualquier edad, y hacerlo en forma esporádica o familiar7,15.
Suele presentarse abruptamente, como evento único o como recidiva, después de un episodio desencadenante como infección, embarazo o exposición a drogas y se manifiesta con AHM, trombocitopenia y daño renal agudo, frecuentemente con hipertensión arterial por sobrecarga de volumen o afectación vascular. Otras veces, los pacientes evolucionan con anemia y trombocitopenia con función renal conservada, o con proteinuria, hipertensión y deterioro progresivo de la función renal2,32,33.
Las manifestaciones extrarrenales dependen del territorio vascular afectado. Son frecuentes las neurológicas, que incluyen irritabilidad, confusión, trastornos motores, convulsiones y coma. También son frecuentes las náuseas, los vómitos y la diarrea, y se han descrito casos con hepatitis, pancreatitis y hemorragia intestinal2. Las manifestaciones cardíacas consisten en enfermedad coronaria con infarto, muerte súbita, cardiomiopatía dilatada y estenosis y trombosis de vasos de mediano calibre en fases tardías de la evolución34,35. También se han descrito casos con afectación pulmonar32 o cutánea36.
Los datos de laboratorio muestran valores de hemoglobina habitualmente inferiores a 10 gm/dl, elevación de LDH y descenso de haptoglobina, en ocasiones hasta valores indetectables. El test de Coombs será negativo, excepto en los casos de infección por neumococo, y en el examen del frotis en sangre periférica se pueden encontrar esquistocitos en cantidades superiores al 1 %, aunque su ausencia no excluye el diagnóstico. La trombocitopenia, cuando se produce, es con frecuencia inferior a 150.000 plaquetas y los estudios de coagulación son normales. El daño renal se objetiva por incremento en los valores de creatinina y por descenso de la tasa de filtrado glomerular o por la presencia de proteinuria y hematuria microscópica. En algunos pacientes pueden encontrarse valores bajos de C3 o C4, aunque su normalidad o la de CFH, CFI, CFB no excluyen la enfermedad.
El análisis genético y funcional del sistema del complemento permite confirmar el diagnóstico y valorar la evolución y el pronóstico. El estudio es imprescindible en los familiares candidatos a donantes vivos para trasplante.
El pronóstico de los pacientes depende del componente del complemento afectado7,32,37 y ha mejorado con el uso de plasmaterapia38 y, sobre todo, de eculizumab. Previamente al uso de eculizumab se producía insuficiencia renal terminal o muerte en el 50-70 % de los casos con mutaciones en CFH, en el 60 % de los que las presentaban en C3, en el 50 % de los pacientes con mutaciones en CFI, CFB y trombomodulina, en el 30-40 % de los portadores de anticuerpos antifactor H y en menos del 10 % en los pacientes con mutaciones en MCP1,39,40.
La recidiva en el trasplante ocurre en más de la mitad de los pacientes con alteraciones de los componentes plasmáticos y es menos frecuente (15-20 %) en los pacientes con alteraciones en MCP, porque la proteína se expresa con normalidad en las células del injerto6,41. La recidiva ensombrece el pronóstico del trasplante, ya que causa pérdida del injerto en el 92 % de los casos6. La mayor edad en la presentación de la enfermedad, un intervalo pequeño entre la entrada en diálisis y el trasplante, el uso de donantes vivos relacionados genéticamente y de anti-calcineurínicos se han considerado como factores de riesgo de recidiva en el trasplante42.
Diagnóstico diferencial
El documento de consenso elaborado por el grupo español para el estudio del SHUa ofrece una excelente estrategia para el diagnóstico diferencial de las MAT en pacientes pediátricos y adultos31. En los adultos es esencial confirmar la AHMA, con o sin trombocitopenia, y evaluar el contexto clínico subyacente incluyendo la gravedad y la evolución del daño renal.
Siempre hay que descartar las enfermedades autoinmunes, la hipertensión maligna, la preeclampsia/HELLP, las infecciones, las neoplasias, los trasplantes de órganos sólidos o hematopoyéticos y el uso de fármacos que puedan inducir daño renal por mecanismos inmunes o por toxicidad dependiente de dosis. Para ello son imprescindibles la historia clínica exhaustiva, el examen físico completo y el uso de las pruebas diagnósticas más sensibles para descartar esos procesos.
Si el daño renal es mínimo o ausente, la púrpura trombocitopénica trombótica (PTT) es la primera opción. Cuando es grave y agudo hay que diferenciar entre PTT y SHU-STEC. Una actividad de ADAMTS13 > 10 % descarta la PTT, mientras que el SHU-STEC se excluye por la ausencia de afectación gastrointestinal y por la negatividad de los cultivos en medio de Mac Conkay para O157:H7, del serotipado específico o de las técnicas de PCR para genes de toxina shiga. La publicación de un caso de SHUa del adulto por déficit de cianocobalamina obliga a considerar este diagnóstico cuando se han descartado el resto de opciones43.
La biopsia renal rara vez es útil para diferenciar las MAT primarias o secundarias, pero puede utilizarse en casos de evolución atípica o para valorar la indicación de tratamiento continuado con plasmaterapia o eculizumab en pacientes con evolución prolongada, sin afectación hematológica y con deterioro grave de la función renal.
TRATAMIENTO
El tratamiento convencional consiste en medidas de soporte y en plasmaterapia (plasmaféresis/infusión de plasma) complementada con inmunosupresión en los pacientes con anticuerpos anti-CFH. Las medidas de soporte incluyen las transfusiones de hematíes cuando la anemia es grave, y de plaquetas si hay riesgo de hemorragia o se realiza alguna intervención, el mantenimiento del volumen intravascular y del soporte nutricional, el control de la hipertensión y el uso de diálisis en caso necesario.
La plasmaterapia aplicada precozmente de forma intensiva y prolongada ha logrado disminuir la mortalidad y alcanzar tasas de remisión hematológica entre 25-88 % según la anomalía presente, pero su eficacia respecto a la función renal es más limitada, porque solo la mitad de los pacientes responden con recuperación o mejoría de la función renal15,38. Además, es ineficaz en los pacientes con alteraciones en MCP, por ser una proteína de membrana, y conlleva complicaciones como la necesidad de un catéter de uso prolongado, la depleción de factores de coagulación o las reacciones anafilácticas, que dificultan su uso continuado.
Para los pacientes con mutaciones en proteínas plasmáticas, como CFH, se han propuesto como soluciones definitivas el trasplante hepático, o el combinado hepático y renal para los que, además, presentan insuficiencia renal terminal. Del análisis de la veintena de casos reportados hasta la fecha se deduce que es imprescindible el uso de plasmaterapia o eculizumab en las fases iniciales del trasplante hasta que la función hepática sea operativa y, en cualquier caso, este tipo de trasplante debe realizarse en centros con experiencia acreditada, después de un cuidadoso balance individualizado de riesgos y beneficios del procedimiento y frente a la alternativa de tratamiento continuado con eculizumab44-47.
El tratamiento del SHUa ha cambiado con el uso de eculizumab, un anticuerpo IgG4/2 kappa monoclonal humanizado con alta afinidad por C548. En adultos, a dosis de 900 mg i.v. semanales durante 4 semanas y de 1.200 mg i.v. cada 2 semanas, posteriormente, bloquea la producción de C5a y de C5-C948,49. Fue aprobado por la FDA (Food and Drug Administration) y la EMA (European Medicines Agency) en 2011, sobre la base de los resultados obtenidos a las 26 semanas de seguimiento en dos estudios observacionales, prospectivos y abiertos, uno con pacientes con SHUa en evolución y refractarios a plasmaterapia, y otro con pacientes de larga evolución, con daño renal crónico y dependientes de plasmaterapia. Los resultados obtenidos con eculizumab en cuanto a abandono de plasmaterapia, normalización hematológica y mejoría de la función renal, sobre todo con su administración precoz, han sido confirmados en esos estudios a más largo plazo50,51, corroborados en otras series, en casos aislados, en una revisión sistemática49,52 y, muy recientemente, en un estudio prospectivo pediátrico53. La eficacia de eculizumab también ha sido demostrada en la profilaxis54-57 y en el rescate de las recidivas en el trasplante renal55,58-60. Sin embargo, la respuesta a eculizumab puede estar limitada en pacientes portadores de mutaciones en C561.
El bloqueo de la vía terminal del complemento favorece la infección por gérmenes encapsulados. Por eso, es preceptiva la vacunación frente a Neisseria meningitidis complementada con profilaxis antibiótica durante el efecto ventana o ininterrumpidamente mientras dure el tratamiento47,62. Por otra parte, el tratamiento continuado con eculizumab es muy costoso en términos monetarios49 y, de momento, se desconocen los criterios y el tipo de monitorización necesarios para su discontinuación individualizada. Las sociedades científicas y los grupos de expertos han elaborado excelentes guías para la optimización de su uso49,31,47. Sin embargo, es preciso continuar investigando para conseguir una metodología sensible y validada para monitorizar la actividad del complemento63-65 y la discontinuación de eculizumab66,67, ya que los estudios en estas áreas son escasos y los resultados no son definitivos.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Correspondencia: Francisco Valdés Cañedo
Hospital Universitario de A Coruña.
As Xubias, 84. 15006 A Coruña.
franvalc@gmail.com


