














INTRODUCCIÓN
En las vasculitis crioglobulinémicas se produce una afectación sistémica de los pequeños y medianos vasos por el depósito de inmunocomplejos que contienen crioglobulinas. Pueden ser primarias o secundarias a diferentes procesos infecciosos, autoinmunes y hematológicas.
Presentamos el caso de una mujer previamente sana, que debutó con síntomas sistémicos inespecíficos, afectación neurológica progresiva y daño renal.
CASO CLÍNICO
Paciente mujer de 45 años, con padre fallecido a los 46 años por complicaciones sistémicas tras un cuadro poliarticular. Antecedentes personales de atopia, episodios de lesiones urticariales y hernia discal L4-L5-S1. Tratamiento ocasional con etoricoxib, dosis puntuales de fosfomicina y furosemida previa al ingreso por edemas declives recientes.
Consultó por cuadro de astenia y debilidad progresiva en los 3 meses previos. Predominaba hipoestesia e hiperalgia en guante y calcetín bilateral, en mayor grado en mano izquierda, con déficit motor grave y febrícula ocasional, con algún episodio de molestias abdominales inespecíficas y autolimitadas.
A su ingreso se objetivó hipertensión grave (164/114 mmHg) y ausencia de edemas. En la exploración neurológica destacaba la abolición del reflejo bicipital y estilorradial izquierdo, con balance motor bilateral disminuido, hipoestesia peroneal superficial, disestesias en ambas manos y fallos de artrocinética.
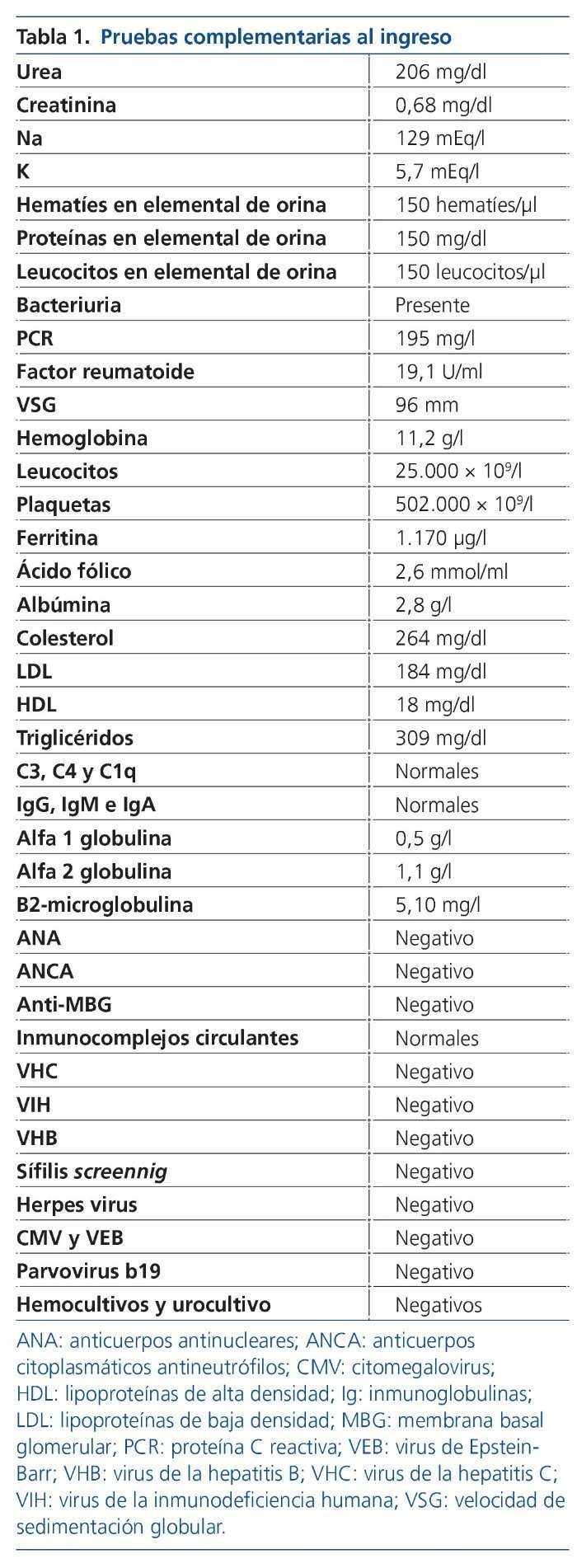
Se inició tratamiento con enalapril para el control tensional, sintomático para el dolor con pregabalina y se comenzó el estudio. Los resultados más significativos se muestran en la tabla 1.
Otras pruebas
• Fondo de ojo, normal.
• Radiografía de tórax, normal.
• Frotis en sangre periférica, con trombocitosis reactiva sin esquistocitos valorables.
• Resonancia magnética craneal y cervical, moderada atrofia corticosubcortical difusa mayor de la esperada para la edad de la paciente, sin otros hallazgos destacables.
• Electroneurograma compatible con mononeuritis múltiple.
• Angio-TC de abdomen y pelvis normal, con riñones de aspecto normal y sin microaneurismas.
• Punción lumbar: líquido cefalorraquídeo (LCR) con 2 células/μl; 12 hematíes/μl; proteínas, 0,15 g/l; glucosa, 0,588 g/l; ADA (adenosina deaminasa), 2,2 U/l. Gram, cultivo y tinción Ziehl-Neelsen sin BAAR (bacilo ácido-alcohol resistente). Citometría de flujo de LCR, sin datos de infiltración por linfoma, sin células neoplásicas y ocasionales linfocitos reactivos.
Evolución inicial
La paciente continuó con progresión de los síntomas neurológicos, deterioro progresivo de la función renal (creatinina, 1,3 mg/dl) y edemas, por lo que se inició tratamiento con metilprednisolona en bolos de 1 g intravenoso (i.v.) durante 3 días y ciclofosfamida 1 g i.v. Se realizaron una biopsia renal percutánea y una del nervio sural.
Resultados anatomopatológicos
• Biopsia del nervio sural. Intensa vasculitis de vaso mediano.
• Biopsia renal.
– Microscopia óptica: 35 glomérulos, con cambios membranoproliferativos con hipercelularidad mesangial y proliferación endocapilar. Fibrosis intersticial/atrofia tubular < 10%. Vasos sin alteraciones relevantes. Seudotrombos hialinos intracapilares. En las figuras de 1 a 5 se muestran imágenes anatomopatológicas de microscopia óptica.
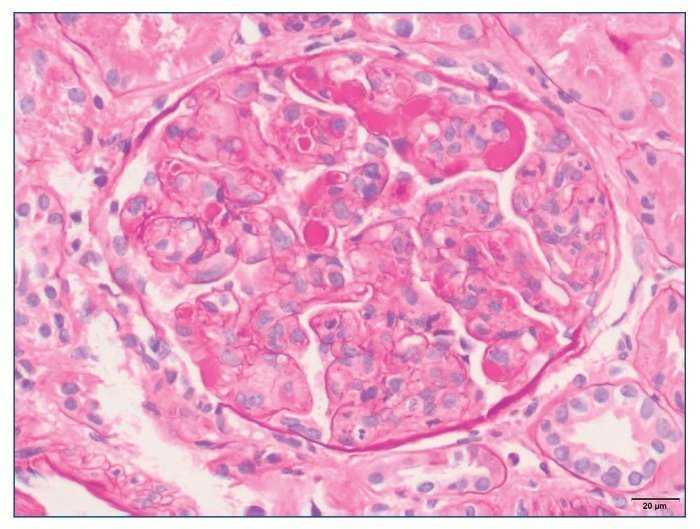
Figura 1. Imagen anatomopatológica de microscopia óptica. Cambios membranoproliferativos.
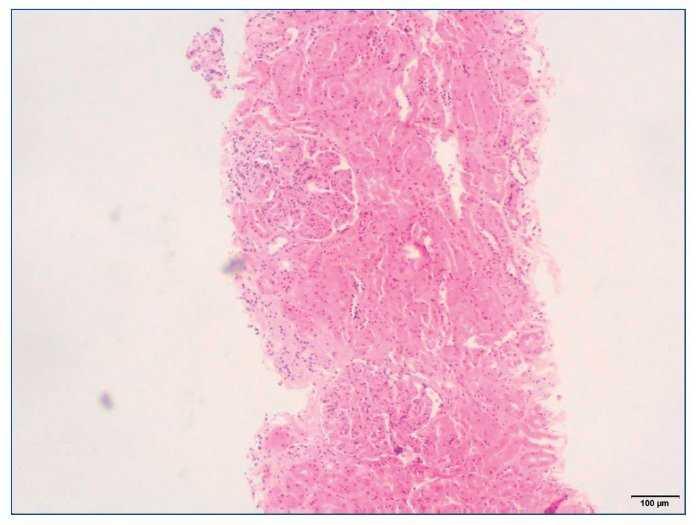
Figura 2. Imagen anatomopatológica de microscopia óptica. Glomérulos hipercelulares.
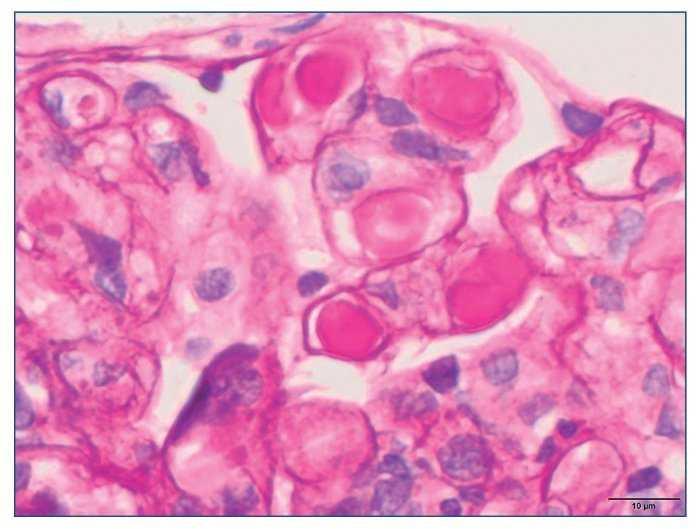
Figura 3. Imagen anatomopatológica de microscopia óptica. Trombos hialinos intraluminales.

Figura 4. Imagen anatomopatológica de microscopia óptica. Trombos hialinos e hipercelularidad mesangial.
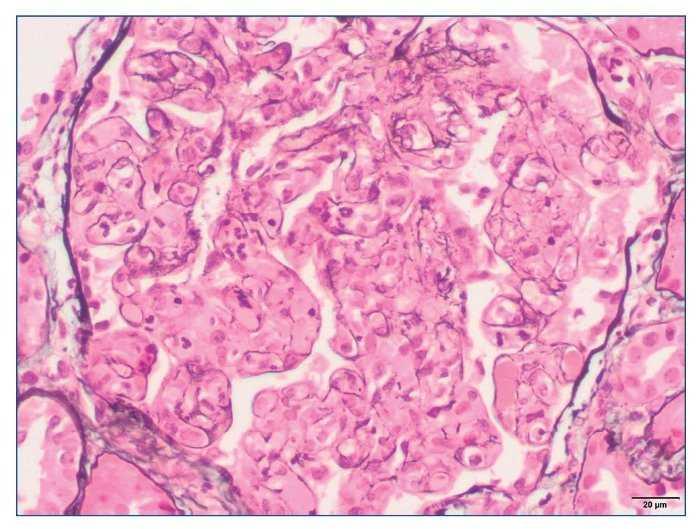
Figura 5. Imagen anatomopatológica de microscopia óptica. Patrón membranoproliferativo.
– Inmunofluorescencia directa (IFD): en la IFD se objetivaron intensos depósitos granulares groseros de IgG e IgM en mesangio, capilares y en seudotrombos hialinos equivalentes a los que se ven en la microscopia óptica, con el mismo patrón e intensidad se observa kappa y en menor medida lambda. Con el mismo patrón se observa también C3. En las figuras 6 a 10 se muestran imágenes anatomopatológicas de IFD.
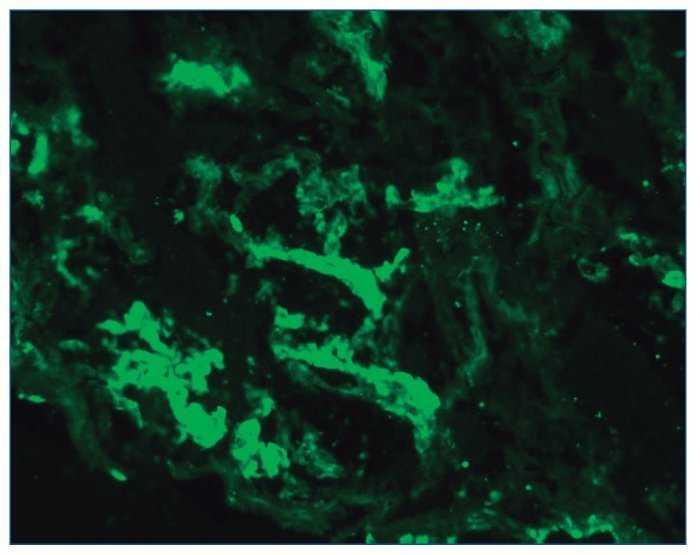
Figura 6. Imagen anatomopatológica de inmunofluorescencia directa. Depósito de IgG.
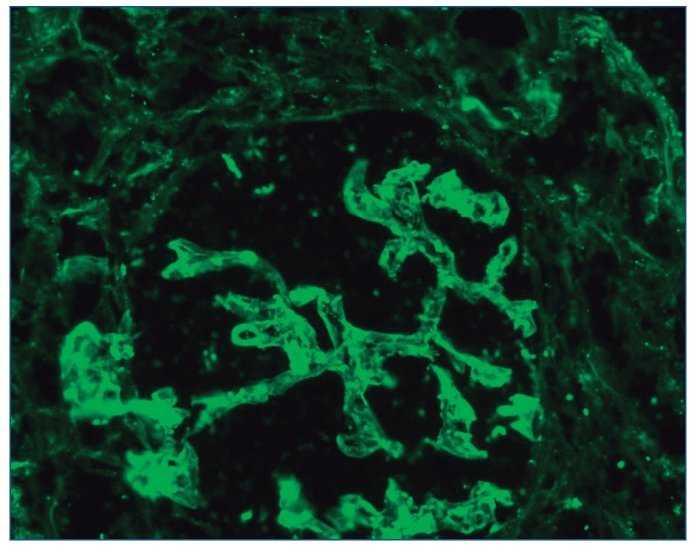
Figura 7. Imagen anatomopatológica de inmunofluorescencia directa. Depósito de IgM.
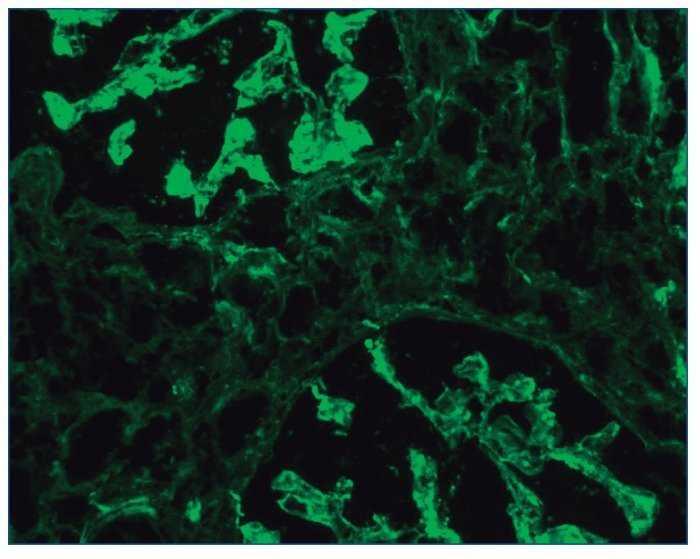
Figura 8. Imagen anatomopatológica de inmunofluorescencia directa. Depósito de cadenas ligeras kappa.
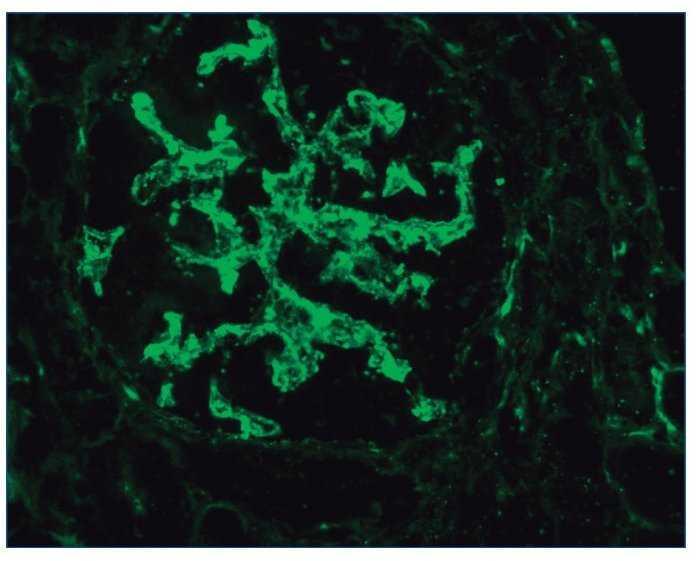
Figura 9. Imagen anatomopatológica de inmunofluorescencia directa. Depósito de C3.
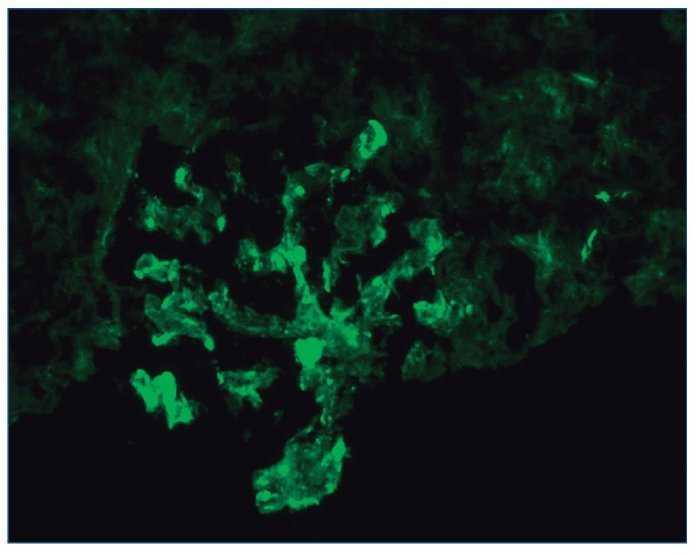
Figura 10. Inmunofluorescencia directa con depósito de C3.
En la figura 11 se muestra el gráfico evolutivo de parámetros analíticos y clínicos: estimación conjunta de la evolución del filtrado glomerular (FG) estimado por CKD-EPI —Chronic Kidney Disease Epidemiology Collaboration— (en ml/min/1,73 m2), proteinuria medida (en mg en 24 h), hematuria medida (en hematíes/µl) y déficit motor y sensitivo evaluado por la exploración en rangos del 0 (ausencia de respuesta motora o sensitiva) al 5 (normalidad).
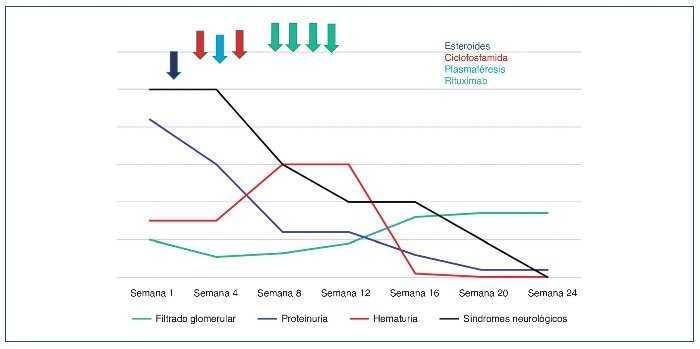
Figura 11. Gráfico evolutivo.
Sospechamos la presencia de una vasculitis sistémica. El diagnóstico diferencial que se planteó inicialmente buscaba descartar procesos autoinmunes por la edad de presentación de la paciente, infecciosos o hematológicos.
Ampliamos las pruebas complementarias solicitadas con este propósito y repetimos algunas para confirmar sus resultados, dada la alta sospecha clínica. En la tabla 2 se muestran los resultado analíticos.
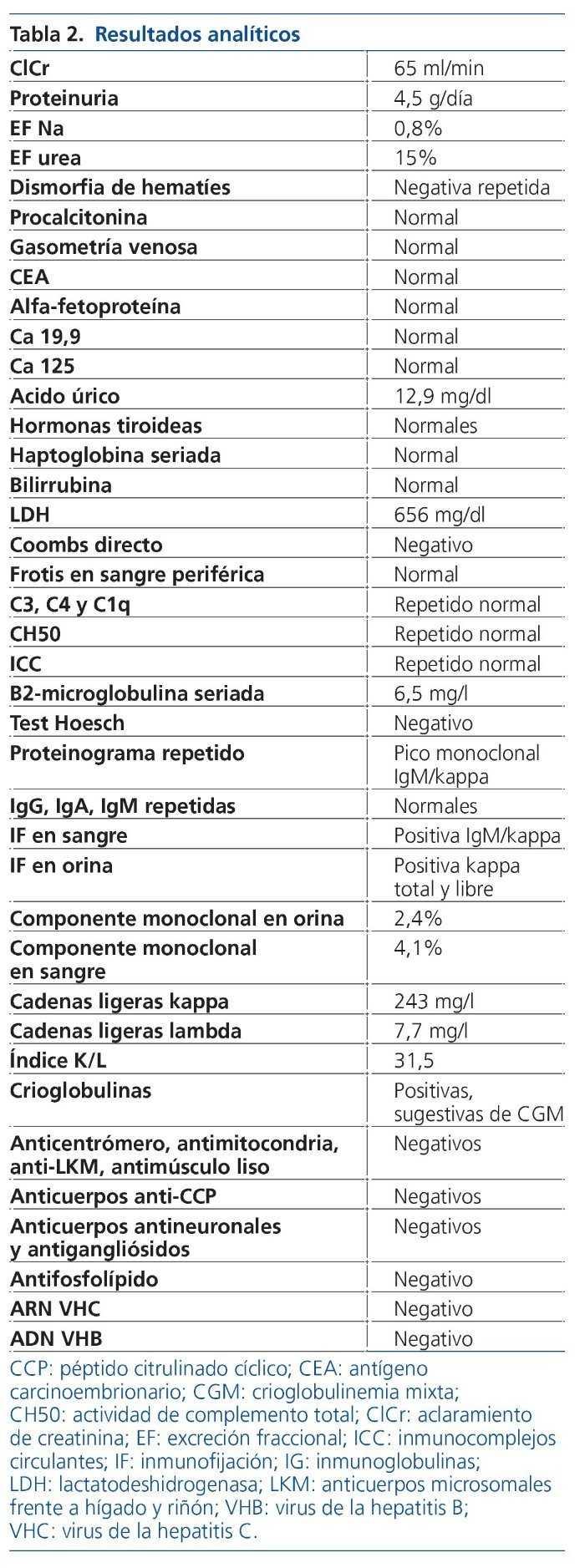
Otras pruebas complementarias
• Estudio de hipercoagulabilidad con leve déficit de proteína S, 44 U/dl (normal, 60-120); DD, 790 µg/l. Consumo de fibrinógeno. Resto, normal.
• PET-TC 4 áreas, normal con baja probabilidad de malignidad.
• Ecocardiograma, normal.
• Eco-Doppler renal, normal.
Ante la probable existencia de una gammapatía monoclonal IgM/kappa, se solicitó estudio de la médula ósea.
Evolución
Se inició plasmaféresis con reposición de albúmina por la rápida progresión de los síntomas neurológicos que mantenían a la paciente postrada, con déficit motor manifiesto y daño renal agudo en progresión. Se realizaron 8 sesiones en total.
Se administró una segunda dosis de 1 g i.v. de ciclofosfamida a los 15 días de la inicial, a la espera del diagnóstico definitivo. Se detectó pancitopenia y alteración de bioquímica hepática en posible relación con toxicidad por la ciclofosfamida, que se recuperó tras la suspensión de esta.
Finalmente, en la citometría de flujo de la médula ósea se objetivó un inmunofenotipo patológico, con un 0,4% de linfocitos B monoclonales kappa. En la biopsia de médula ósea había abundante celularidad medular con hematopoyesis bien representada con signos inespecíficos de reactividad, pero aún sin datos de infiltración medular y sin criterios diagnósticos claros de enfermedad de Waldeström.
Se decidió, de forma consensuada con hematología y neurología, el inicio de tratamiento con rituximab a dosis habitual de 375 mg/m2 i.v. semanal durante 4 semanas más de forma ambulatoria, como indicación tras el diagnóstico definitivo de vasculitis secundaria a crioglobulinemia tipo 2 asociada a gammapatía monoclonal IgM/kappa.
Tras el inicio de inmunomoduladores y plasmaféresis, la paciente comenzó a mejorar clínicamente, con lenta recuperación del déficit neurológico, mejoría de la función renal y reducción de la proteinuria, hematuria y leucocituria. Se logró la resolución absoluta de los síntomas neurológicos. En la actualidad se encuentra en remisión completa hematológica, neurológica y renal, con normalización del aclaramiento de creatinina, sedimento urinario y desaparición de proteinuria significativa. La desaparición de los datos de afectación renal se produjo a los 2 meses de haber iniciado el tratamiento inmunomodulador. El descenso progresivo de esteroides finalizó a los 6 meses.
DISCUSIÓN
La vasculitis crioglobulinémica se produce por depósitos de complejos inmunes en vasos de pequeño y mediano tamaño. Estos inmunocomplejos están formados por inmunoglobulinas que precipitan a menos de 37 ºC y se vuelven a disolver al calentarlas.
La clasificación de Brouet las divide en 3 tipos:
• Tipo I, es la más infrecuente, se detecta un componente monoclonal de cualquiera de las inmunoglobulinas o cadenas ligeras libres. Se asocia a gammapatías de significado incierto, mieloma múltiple, Waldeström y leucemia linfática crónica.
• Tipo II, la más frecuente, la encontramos en más del 60% de todas las vasculitis crioglobulinémicas. En el crioprecipitado se detectan IgM monoclonal e IgG de carácter policlonal. Se socia a infecciones virales, sobre todo al virus de la hepatitis C (VHC), pero también al virus de la hepatitis B (VHB), al virus de la inmunodeficiencia humana (VIH) y al virus de Epstein-Barr (VEB). Se puede manifestar en procesos autoinmunes como el lupus eritematoso sistémico (LES) o en cuadros linfoproliferativos. En el 10% de los casos no se encuentra causa asociada y se catalogan como primarias.
• Tipo III, presente en algo más de un tercio de los casos. En este tipo se encuentran IgM e IgG, pero ambas policlonales. Suelen asociarse a enfermedades autoinmunes o al VHC. Rara vez son primarias.
Los tipos II y III son las denominadas crioglobulinemias mixtas.
Epidemiológicamente se asocian mayoritariamente con el VHC, sobre todo en casos evolucionados y en fase de cirrosis. Aproximadamente, en el 25% de las enfermedades autoinmunes o de los procesos linfoproliferativos puede aparecer crioglobulinemia. La mayoría no desarrolla vasculitis. Por otra parte, hasta en un 40% de los síndromes de crioglobulinemia mixta no se logran detectar crioglobulinas.
La presentación clínica típica en el tipo I depende de la oclusión vascular por el crioprecipitado. En las crioglobulinemias mixtas, es típica la afectación fluctuante con síntomas constitucionales poco específicos, púrpura palpable, debilidad y neuropatía periférica, con afectación renal en hasta un 30% de los casos. Es típica la tríada de Meltzer, que incluye púrpura, artralgia y mialgias.
Las manifestaciones renales suelen aparecer a los 2-3 años del diagnóstico. Típicamente se presentan con hipertensión arterial, hematuria y proteinuria, en casos raros como glomerulopatía rápidamente progresiva o, en el otro extremo del espectro, como enfermedad renal crónica sin otras anormalidades y sin afectación sistémica. Globalmente aparecen en el 20% de los casos de vasculitis crioglobulinémicas, sobre todo como afectación por inmunocomplejos en los casos de crioglobulinemia mixta con patrón membranoproliferativo. En los casos asociados al tipo I, predomina el daño por patología trombótica.
Debemos sospecharla en los casos con artralgias, púrpura, úlceras, glomerulopatías y neuropatía periférica motora que acompaña a síntomas constitucionales. Nos obliga a descartar procesos virales, neoplasias hematológicas o enfermedades autoinmunes. Pueden orientar el diagnóstico el complemento C4 bajo, el factor reumatoide positivo, la velocidad de sedimentación globular (VSG) y la proteína C reactiva (PCR) alta, la hiper-gammaglobulinemia y la presencia de leucocitosis y trombocitosis.
La anatomía patológica en piel suele manifestarse como vasculitis leucocitoclástica, como trombos hialinos o como secuelas postinflamatorias en las mixtas. En el tipo I son más frecuentes los fenómenos trombóticos.
En el nervio periférico, en el tipo I, son típicas las lesiones oclusivas. En las mixtas se presenta vasculitis necrosante de vasos epineurales. En los resultados del electromiograma es muy frecuente hallar mononeuritis múltiple.
Respecto a las lesiones típicas en la biopsia renal, en el 60-80% de los casos se presenta como glomerulopatía membranoproliferativa, con engrosamiento de la membrana basal glomerular, proliferación endocapilar y depósitos subendoteliales e intraluminales de crioglobulinas, otras inmunoglobulinas y proteínas del complemento. Al microscopio óptico son típicos los trombos intraluminales.
En el 20-30% se presentan como una glomerulopatía proliferativa mesangial con trombos hialinos intraglomerulares y vasculitis con necrosis fibrinoide.
En el tipo I, como a otros niveles sistémicos, predominan las lesiones isquémicas e hipocelulares no inflamatorias.
Al microscopio electrónico son típicos los depósitos granulares subendoteliales electrodensos, como huellas dactilares.
El tratamiento de elección en casos de afectación sistémica progresiva y casos graves son los esteroides en ciclo corto (sobre todo en los casos de afectación viral), el rituximab como fármaco de elección y el uso adyuvante de plasmaféresis en casos seleccionados.
Se consideran casos graves los que presentan glomerulopatía rápidamente progresiva, isquemia digital grave, vasculitis intestinal o hemorragia digestiva, neuropatía progresiva, accidente cerebrovascular agudo o déficit de funciones superiores, vasculitis pulmonar, insuficiencia cardíaca o hiperviscosidad.
Una vez estabilizados, el tratamiento debe ser el de la enfermedad subyacente. Hasta el 60% de los casos mejoran entre 12 y 24 meses.
El diagnóstico diferencial de esta entidad incluye:
• Vasculitis de pequeño y mediano vaso: drogas, glomerulopatía IgA, vasculitis asociadas a ANCA, asociadas a infecciones.
• Vasculitis asociadas a enfermedades del tejido conectivo: LES, síndrome de Sjogren, artritis reumatoide.
• Simuladoras: infecciones, enfermedades tromboembólicas (síndrome antifosfolípido, anemia falciforme, púrpura trombótica trombocitopénica, síndrome hemolítico urémico, mixoma auricular).
• Otras causas de descenso de perfusión celular: policitemia, parasitosis (malaria), glomerulopatías con otras Ig no crioglobulinas.
La supervivencia global es del 70% a los 10 años del inicio de los síntomas y del 50% a los 10 años del diagnóstico.
Hasta el 50% de los casos con afectación renal pueden remitir con tratamiento a los 6 meses. Recurren del 50 al 70% tras el trasplante, pero no suelen perder el injerto por este motivo.
En nuestro caso, la clínica, con molestias abdominales inespecíficas, astenia progresiva, debilidad, afectación neurológica y daño renal con hipertensión, nos hizo sospechar desde el inicio el posible diagnóstico de vasculitis sistémica. Una vez descartadas las enfermedades autoinmunes y la presencia de virus, nos decantamos por insistir en la búsqueda de una posible alteración hematológica asociada. Los cambios membranoproliferativos, la proliferación endocapilar y la típica presencia de seudotrombos hialinos intracapilares, así como los depósitos predominantes de cadena ligera kappa, nos apoyaron en el diagnóstico de presunción.
De forma retrospectiva, nos faltaban algunos síntomas cardinales, como las lesiones cutáneas en forma de púrpura o isquemia, o las artralgias. El momento de presentación tampoco era el típico sin diagnóstico previo de enfermedad. Algunos datos de laboratorio no apoyaban el diagnóstico, como la normalidad del C4 e inmunocomplejos circulantes, la ausencia de dismorfia de los hematíes en el sedimento y un primer proteinograma normal. La presencia de crioglobulinas en este contexto y la detección de la gammapatía monoclonal IgM kappa en el segundo proteinograma con el estudio de médula ósea confirmatorio nos dieron la clave. La dificultad en la correcta extracción y transporte de las crioglobulinas, en este caso solo débilmente positivas, no es infrecuente y también puede ser un factor de confusión.
Agradecemos los comentarios recibidos. Son muy acertados y plantean perfectamente el diagnóstico diferencial. Probablemente en la descripción de la inmunofluorescencia directa no
hemos dejado suficientemente claro el predominio de las cadenas ligeras kappa. Por otro lado, la indicación de plasmaféresis queda como tema para discusión. En nuestro caso, la paciente presentaba una rápida progresión de los síntomas neurológicos motores, lo que nos hizo indicarla. Es difícil saber si hubiera mejorado de igual forma solo con esteroides y rituximab, pero posteriormente nos llamó la atención la correlación en el tiempo de la mejoría neurológica y el inicio de la plasmaféresis.
CONCLUSIONES
La crioglobulinemia mixta es una entidad que se debe sospechar en los casos que se presentan con artralgias, púrpura o lesiones isquémicas, glomerulopatía y neuropatía periférica motora que acompañan a síntomas constitucionales. La lesión típica es la vasculitis de vasos pequeño y mediano.
La crioglobulinemia tipo 2 (Ig monoclonal IgM y policlonal IgG) es la que más frecuentemente se asocia a glomerulopatías, relacionándose frecuentemente con el VHC, disproteinemias, infecciones y enfermedades autoinmunes. En este caso hemos descartado el tipo 1 por aparición en la IFD en la biopsia renal de IgG e IgM, y el tipo 3 por estar menos frecuentemente implicada en el desarrollo de glomerulopatías y por el componente monoclonal IgM.
La ausencia de algunos de los síntomas típicos o la negatividad de algunas pruebas complementarias en ocasiones nos confunden, en algunos casos por variabilidad dependiente del observador y en otros simplemente por estar ausentes.
La afectación sistémica, no solo renal, asociada a enfermedades hematológicas ocultas posiblemente esté infraestimada y habría que pensar en esta posibilidad en los casos clínicamente compatibles.
Conflicto de intereses
Los autores declaran que no tienen conflicto de intereses relacionado con los contenidos de este artículo.
Correspondencia:
Macarena Naranjo Arellano
Servicio de Nefrología.
Hospital Universitario Virgen del Rocío. 41013. Sevilla.
macarena.naranjo.sspa@juntadeandalucia.es
Revisión por expertos bajo la responsabilidad de la Sociedad Española de Nefrología.


