



La poliquistosis renal autosómica dominante (PQRAD) es una de las principales causas de insuficiencia renal terminal. Hoy día el conocimiento de sus bases genéticas está permitiendo desarrollar estrategias que eviten la transmisión de la enfermedad.
ObjetivosEl objetivo del estudio fue analizar la historia natural de la PQRAD en la provincia de Córdoba y diseñar una base de datos que permita agrupar a las familias y a las diferentes mutaciones.
Pacientes y métodosSe incluyen todos los pacientes (n=678) diagnosticados de PQRAD seguidos en el servicio de Nefrología de Córdoba. Se analizaron de manera retrospectiva diversas variables clínicas (edad y sexo), necesidad de terapia renal sustitutiva (TRS) y genéticas.
ResultadosLa prevalencia fue de 61 casos por 100.000 habitantes. La mediana de supervivencia renal fue significativamente peor en PKD1 (57,5 años) que en PKD2 (70 años) (Log-rank p=0,000). Tenemos identificada genéticamente al 43,8% de la población, detectando mutaciones en PKD1 en el 61,2% y en PKD2 en el 37,4% de los casos. La mutación más frecuente fue detectada en PKD2 (c.2159del) en 68 pacientes pertenecientes a 10 familias diferentes. La de peor pronóstico renal fue una mutación truncante detectada en PKD1 (c.9893G>A).
ConclusionesLa supervivencia renal de la PQRAD en la provincia de Córdoba es similar a la descrita en la literatura. Con nuestra metodología y estudiando genéticamente al 43,8% de la población, detectamos mutaciones en PKD2 en una mayor proporción de pacientes, en el 37,4% de los casos. Esta estrategia permite conocer las bases genéticas de gran parte de nuestra población con ahorro de recursos. Esto es fundamental para poder ofrecer prevención primaria de la PQRAD mediante diagnóstico genético preimplantacional.
Autosomal dominant polycystic kidney disease (ADPKD) is one of the main causes of end-stage renal disease. Today, the knowledge of its genetic base has made it possible to develop strategies that prevent the transmission of the disease.
ObjectivesThe objective of the study was to analyze the natural history of ADPKD in the Province of Córdoba and to design a database that allows families and different mutations to be grouped.
Patients and methodsAll patients (n=678) diagnosed with ADPKD followed up in the Cordoba nephrology service are included. Various clinical variables (age and sex), genetic variables (mutation in PKD1, PKD2) and the need for renal replacement therapy (RRT) were retrospectively analyzed.
ResultsThe prevalence was 61 cases per 100,000 inhabitants. Median renal survival was significantly worse in PKD1 (57.5 years) than in PKD2 (70 years) (Log-rank p=0.000). We have genetically identified 43.8% of the population, detecting mutations in PKD1 in 61.2% and in PKD2 in 37.4% of cases. The most frequent mutation was detected in PKD2 (c.2159del) in 68 patients belonging to 10 different families. The one with the worst renal prognosis was detected in PKD1 (c.9893G>A). These patients required RRT at a median age of 38.7 years.
ConclusionsThe renal survival of ADPKD in the Province of Córdoba is similar to that described in the literature. Mutations in PKD2 were detected in 37.4%. Our strategy allows us to know the genetic bases of our population with a great saving of resources. This is essential to be able to offer primary prevention of ADPKD through preimplantation genetic diagnosis.
La poliquistosis renal autosómica dominante (PQRAD) es la enfermedad renal hereditaria más frecuente, con una prevalencia global de 1 cada 1.000 personas, siendo una de las primeras causas de insuficiencia renal terminal (IRT)1–3 en todo el mundo. La manifestación clínica más característica es la formación de quistes renales que a lo largo de la vida van aumentando en número y tamaño. El parénquima renal normal se sustituye por estos quistes, por lo que a partir de la 2.ª-5.ª década aparece litiasis, hipertensión arterial, hematuria e insuficiencia renal, la manifestación más severa de la enfermedad. El 50% de los pacientes con PQRAD precisan terapia renal sustitutiva (diálisis o trasplante) a una edad media de 57 años4,5.
Es una enfermedad genética autosómica dominante6 con penetrancia completa. Es una enfermedad heterogénea, en la que los principales genes están bien identificados: PKD1, en el cromosoma 16p13.3 y PKD2, en el cromosoma 4q21-237,8. La proporción de mutaciones se describen en un 85% de los casos en PKD1 y solamente un 15% en PKD27,8, aunque esta proporción podría variar según el área geográfica y la proporción de pacientes estudiados. Los individuos con mutaciones en PKD1 tienden a tener una presentación clínica más severa, aunque con una gran variabilidad interfamiliar e intrafamiliar. La mayoría de los individuos con mutaciones en PKD1 desarrollan IRT a una edad media de 54,3 años; por el contrario, más de un 50% de los individuos con mutaciones en PKD2 tienen una adecuada función renal a dicha edad (edad mediana de IRT, 74,0 años)9,10.
La ecografía renal es el estudio de imagen habitualmente empleado debido a su inocuidad y a su bajo coste. Actualmente, los criterios ecográficos de Ravine modificados son los más aceptados11. El desarrollo de los quistes renales en la PQRAD comienza desde la etapa embrionaria; estos quistes continúan aumentando de tamaño durante la vida del individuo. El estudio «Consortium of imaging studies to assess the progression of polycystic kidney disease» (CRISP)12,13 es el que ha proporcionado la mejor información clínica acerca del crecimiento renal y pronóstico renal a largo plazo.
El desarrollo de insuficiencia renal es muy variable, como ya hemos comentado anteriormente, y depende principalmente de la presencia del gen PKD1 o PKD2. Otros factores que influyen en el curso clínico de la enfermedad incluyen el sexo masculino, diagnóstico antes de los 30 años, primer episodio de hematuria antes de los 30 años, inicio de hipertensión antes de los 35 años, hiperlipemia y colesterol HDL bajo14. La PQRAD es una enfermedad multisistémica, donde además de quistes renales, existen otras manifestaciones extrarrenales; quistes hepáticos, anomalías vasculares15, cardíacas, digestivas y musculoesqueléticas16. La enfermedad poliquística hepática (EPQH) es la manifestación extrarrenal más frecuente y se asocia con ambos genotipos: PKD1 y PKD2. Actualmente el único tratamiento que ha demostrado cierto beneficio en retrasar el deterioro de la enfermedad renal crónica es el tolvaptán17.
Es fundamental conocer las bases genéticas de las enfermedades, sobre todo teniendo en cuenta que hoy día es posible evitar la transmisión primaria de ellas mediante el diagnóstico genético preimplantacional (DGP). Todos los pacientes con PQRAD deberían tener un estudio genético y ofertar el DGP antes de la edad reproductiva. Los estudios genéticos son caros y no están indicados actualmente de manera rutinaria en todos los pacientes, así pues, es necesario diseñar estrategias que con el menor coste económico posible obtengan mayores resultados en salud.
El objetivo de nuestro estudio fue analizar la historia natural de la PQRAD en la provincia de Córdoba. Para ello diseñamos una base de datos que nos permitiera agrupar a las familias y a las diferentes mutaciones con la idea de rentabilizar los estudios genéticos y ofrecer prevención primaria a nuestra población.
Pacientes y métodosPara este estudio se ha utilizado una base de datos que incluye todos los pacientes con PQRAD seguidos en el servicio de Nefrología de Córdoba. Esta base se actualiza periódicamente, la última actualización se realizó en enero de 2021. Los pacientes se incluyen si cumplen los criterios de Ravine modificados10 o son portadores de una mutación genética patogénica.
VariablesSe analizaron las siguientes variables: fecha de nacimiento, sexo, fecha de terapia renal sustitutiva (TRS), fecha de fallecimiento y estudio genético. Los estudios genéticos se realizaron mediante el método de NGS (Next-Generation Sequencing) para los genes PKD1, PKD2 y GANAB. Adicionalmente, se utilizó la técnica MLPA (Multiplex ligation-dependent probe amplification) para deleciones/duplicaciones de PKD1 y PKD2, cuando estuvo indicada.
Se consideró una mutación como responsable de la enfermedad cuando era patogénica, probablemente patogénica o la familia presentaba un estudio de segregación. Los pacientes se agruparon según el gen afecto (PKD1, PKD2), la mutación concreta y la familia a la que pertenecían.
Se ha realizado un estudio descriptivo de las variables cuantitativas y cualitativas. Se calculó la supervivencia renal y del paciente. Todos los contrastes son bilaterales y hemos considerado como significativos aquellos donde p<0,05. Los datos han sido recogidos, procesados y analizados con el programa estadístico SPSS v.17.
Aspectos éticosLos datos se manejaron de manera anonimizada, respetando la ley de protección de datos. Se respetaron todos los requerimientos éticos y legales promovidos en nuestro país para asegurar tanto la confidencialidad como la buena práctica en materia de investigación clínica aplicada. Así mismo, se han cumplido en todo momento los preceptos éticos contenidos en la Declaración de Helsinki.
ResultadosEstudio descriptivoEn el periodo de tiempo estudiado (1990-2021), han sido diagnosticados 678 enfermos con PQRAD en la provincia de Córdoba. De nuestra muestra, 350 eran mujeres (51,6%) y 328 varones (48,4%). En este periodo han fallecido 197 (29,1%). En enero de 2021, 481 pacientes se seguían en el servicio de Nefrología de Córdoba. Considerando que según el Instituto Nacional de Estadística (INE) la población de Córdoba el 1 de enero de 2020 era de 784.256 habitantes, la prevalencia de PQRAD en la provincia de Córdoba es de 61/100.000 habitantes.
De estos 481, 168 (34,9%) están en TRS. De estos 168 pacientes en TRS, 50 (29,7%) están en las diferentes técnicas de diálisis y 118 (70,2%) presentan un trasplante renal funcionante. Por otro lado, 313 pacientes (65,1%) se siguen en consultas externas de nefrología. La edad media de los pacientes en TRS fue de 53,0±11,6 frente a 51,6±16,1 años (p<0,0001) de los pacientes sin TRS seguidos ambulatoriamente.
Supervivencia renal y del pacienteLa mediana de supervivencia renal para toda la cohorte de 678 pacientes fue de 63,2 años. La supervivencia renal fue mayor en mujeres que en hombres, 64,3 vs. 61,0 años (Log-rank, p=0,011) (fig. 1). La mediana de supervivencia del paciente fue de 78,9 años. En hombres fue de 75,6 años y en la mujer de 81,04 años (Log-rank, p=0,026).
Estudio genético
Tenemos clasificados genéticamente a 297 de los 678 pacientes (43,8%) por realizarse estudios genéticos o pertenecer a familias con una mutación patogénica, probablemente patogénica o con estudio de segregación identificado. Solo 381 pacientes no han sido estudiados o clasificados genéticamente (56,2%). De los 297 estudiados genéticamente, se encontró mutación en PKD1 en 182 (61,2%), en PKD2 en 111 (37,4%) y 2 en gen GANAB (gen que codifica la subunidad alfa II glucosidasa) (0,7%). Detectamos 74 variantes genéticas en nuestra población, siendo 43 (58,1%) patogénicas, 22 (29,7%) probablemente patogénicas y 9 (12,2%) de significado incierto. De las 9 variables de significado incierto, una estaba descrita en las bases de datos y las 8 restantes no.
La mediana de supervivencia renal para PKD1 fue de 57,5 años y de 70 años para PKD2 (Log-rank p=0,000) (fig. 2). La mediana de supervivencia renal para los pacientes no identificados genéticamente (n=381) fue de 62,7 años.
Agrupación familiar y según la mutación
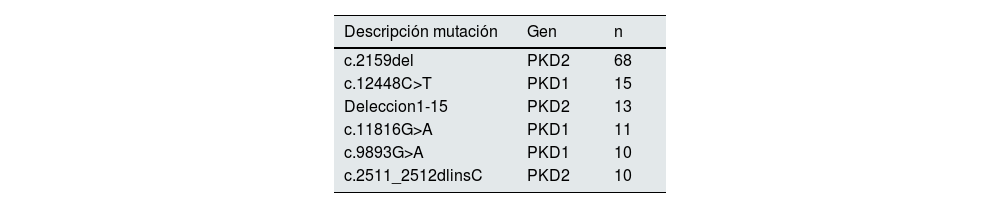
Los estudios genéticos se realizaron en 100 familias diferentes y se identificaron 62 mutaciones diferentes. En la tabla 1 se presentan las mutaciones que se detectaron con más frecuencia. Se presentan solo mutaciones en las que se detectaron al menos 10 casos. La más frecuente, que se presentó en 68 casos, fue la mutación truncante c.2159del en PKD2.
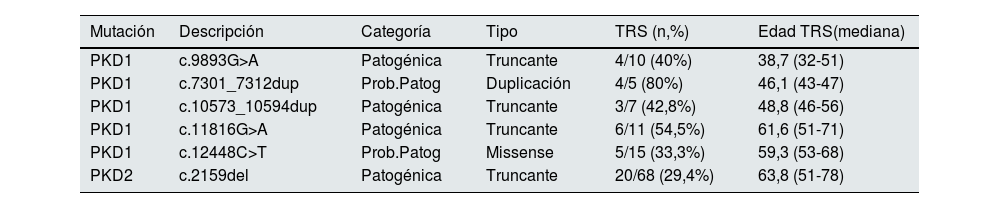
En la tabla 2 se presentan las mutaciones que se asociaron al desarrollo de enfermedad renal crónica terminal con mayor severidad. Se describen solo aquellas mutaciones en las que se identificaron al menos 3 miembros con necesidad de TRS. Podemos observar que, de estas 6 mutaciones, 5 pertenecen a PKD1 y una a PKD2. También que 4 de las 6 son mutaciones truncantes.
Mutaciones con peor pronóstico renal
| Mutación | Descripción | Categoría | Tipo | TRS (n,%) | Edad TRS(mediana) |
|---|---|---|---|---|---|
| PKD1 | c.9893G>A | Patogénica | Truncante | 4/10 (40%) | 38,7 (32-51) |
| PKD1 | c.7301_7312dup | Prob.Patog | Duplicación | 4/5 (80%) | 46,1 (43-47) |
| PKD1 | c.10573_10594dup | Patogénica | Truncante | 3/7 (42,8%) | 48,8 (46-56) |
| PKD1 | c.11816G>A | Patogénica | Truncante | 6/11 (54,5%) | 61,6 (51-71) |
| PKD1 | c.12448C>T | Prob.Patog | Missense | 5/15 (33,3%) | 59,3 (53-68) |
| PKD2 | c.2159del | Patogénica | Truncante | 20/68 (29,4%) | 63,8 (51-78) |
PKD1 y 2: Polycistic Kidney Disease 1 y 2; TRS: terapia renal sustitutiva; n: número de pacientes.
La mutación de peor pronóstico de nuestra cohorte se localiza en PKD1, c.9893G>A. El 40% de los pacientes de esta familia necesitan TRS a una edad mediana 38,7 años (rango 32-51).
Agrupando los casos por mutaciones y familias hemos observado la misma mutación en 68 miembros pertenecientes a 10 familias. Es una mutación truncante en PKD2 (c.2159del) ya descrita y considerada patogénica. El 29,4% de los pacientes con esta mutación precisaron TRS a una edad media de 65,2±7,6 años. Esta mutación se agrupa en un área geográfica concreta de Córdoba.
DiscusiónEste estudio surgió de la necesidad de conocer la epidemiología, distribución e historia natural de la PQRAD en la provincia de Córdoba. También de la necesidad de diseñar estrategias sanitarias que actúen eficientemente ante esta enfermedad centradas en su estudio genético. Este estudio tiene la fortaleza que incluye a todos los pacientes diagnosticados de PQRAD en nuestra provincia, por lo que nos permite hacer estimaciones muy exactas de la prevalencia, distribución de las mutaciones e historia natural.
La PQRAD es la enfermedad hereditaria más frecuente con una prevalencia descrita de 1/1000-2000 habitantes (50-100/100.000). El estudio más riguroso, realizado en 2020 por la Clínica Mayo (Condado de Olmsted), cifró la prevalencia de la enfermedad «definitiva/probable» en 68/100.000 habitantes18. Estos autores especulan que el diagnóstico de enfermedad «posible» de PQRAD puede llegar hasta 234/100.000 habitantes. En nuestro estudio, la prevalencia fue muy parecida, de 61/100.000. En Andalucía, un análisis realizado en Granada19 cifró la prevalencia en 58/100.000 habitantes. Conocer estos datos es fundamental para programar las necesidades sanitarias de esta población, sobre todo de estos casos más graves. Según el sistema de información de la Coordinación Autonómica de Trasplantes de Andalucía (SICATA), más del 10% de todos los pacientes en TRS (diálisis y trasplante) en Andalucía lo están por padecer una PQRAD. El coste anual de estos pacientes (1068 en el año 2019) puede oscilar entre 30-50 millones de euros. Son necesarias estrategias que ofrezcan a estas familias y al sistema sanitario un abordaje que incluya la prevención primaria de la enfermedad con las técnicas de DGP20. Esto es especialmente importante en nuestro medio ya que estas técnicas no están llegando efectivamente a la población que lo necesita.
Es una enfermedad genética autosómica dominante causada principalmente por mutaciones en 2 genes, PKD1 y PKD221. Las mutaciones en el gen PKD1 se identifican en todos los estudios con mayor frecuencia (85%) que las mutaciones en PKD2 (15%)22. Probablemente estos valores reflejan la población estudiada genéticamente, no la prevalencia real de cada una de ellas. Los pacientes con mutaciones en PKD1 tienen una enfermedad más severa y por tanto suelen estudiarse genéticamente en mayor proporción que los pacientes con mutaciones en PKD2. Cornec-Le Gall et al.23, en un estudio multicéntrico de 22 centros, que incluyó 741 pacientes, detectaron mutaciones en PKD1 en el 75,5% de los pacientes y en PKD2 en el 18,3%. En nuestro estudio que incluyó a 678 pacientes de un único centro, detectamos mutaciones en PKD1 en el 61,2% de los pacientes y en PKD2 en el 37,4%. Esta diferencia podría deberse a nuestra estrategia, que ha conseguido identificar genéticamente al 43,8% de los casos. Tampoco se puede descartar que las mutaciones en PKD2 sean más frecuentes en nuestra provincia. Apoyaría esta hipótesis el hecho de haber detectado un gran número de casos (n=68) en una zona geográfica concreta que sugiere claramente una mutación con efecto fundador. Solamente podremos contestar a esta pregunta cuando en la mayoría de los centros tengamos identificada a la mayoría de la población. Evidentemente hay otros genes minoritarios y algunos todavía no descritos24. En 2 casos detectamos mutaciones en el gen GANAB (0,7%) y en 2 casos no detectamos ninguna mutación.
El pronóstico renal depende de la mutación, los pacientes con mutaciones en PKD1 presentan IRC terminal a una edad mediana de 54,3 años (frente a 74 años en PKD2). Nuestros datos son superponibles, la mediana de supervivencia renal para PKD1 fue de 57,5 años y de 70 años para PKD2 (Log-rank, p=0,000). Como en la mayoría de los estudios, la supervivencia del paciente fue significativamente mayor en mujeres que en hombres (75,6 vs. 81,0, p=0,026). A destacar quizás el dato de que la supervivencia renal también fue significativamente mejor en mujeres que en hombres (64,3 vs. 61,0 (Log-rank, p=0.011). Esto no debe extrañar ya que el sexo masculino es factor de riesgo de progresión de la enfermedad renal en la mayoría de las nefropatías y también se está describiendo recientemente en pacientes con PQRAD25.
Con nuestra estrategia para agrupar mutaciones y familias identificamos 74 mutaciones en 100 familias no relacionadas. Esto permite una vez identificada la familia no repetir estudios genéticos en sus miembros. Es curioso que hemos observado la misma mutación en 68 miembros pertenecientes a 10 familias diferentes que se concentran en un área geográfica de Córdoba. Esta es una mutación patogénica truncante en PKD2. En ella se produce una deleción de una adenina (c.2159del) que da lugar a un codón de parada prematuro. También hemos identificado las mutaciones con peor pronóstico renal. Como era de esperar, 5 de las 6 eran en PKD1 y 4 de las 6 eran truncantes. La más severa (edad mediana de TRS de 38,7 años) se localizaba en PKD1. En esta familia se identificó un cambio en el nucleótido (c.9893G>A) en heterocigosis en el exón 29 del gen PKD1. Esta mutación da lugar a una proteína de 3298 aminoácidos en vez de los 4302 de la proteína nativa.
En resumen, la supervivencia renal de la PQRAD en la provincia de Córdoba es similar a la publicada en la literatura, siendo significativamente peor en PKD1 (57,5 años) que en PKD2 (70 años). La prevalencia de PQRAD fue de 61/100.000. Diseñamos una estrategia para agrupar familias y mutaciones identificando genéticamente al 43,8% de la población, detectando mutaciones en PKD1 en el 61,2% y en PKD2 en el 37,4 de los casos. Identificamos las mutaciones más frecuentes en nuestra población y aquellas con peor pronóstico renal. Esta estrategia puede ser muy útil para el abordaje futuro mediante técnicas de reproducción humana en esta enfermedad tan frecuente y severa.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



