

La hipouricemia renal hereditaria es un trastorno genético, poco frecuente, causado por un defecto aislado en la reabsorción del ácido úrico a nivel del túbulo renal. Los pacientes presentan concentraciones séricas de ácido úrico inferiores a 2 mg/dl (119 micromol/L), y un incremento en la excreción fraccional de ácido úrico mayor del 10%. La mayoría son asintomáticos y se detectan accidentalmente, aunque pueden aparecer complicaciones como la nefrolitiasis, hematuria, daño renal agudo inducido por ejercicio físico o tras un episodio de deshidratación por gastroenteritis aguda, o el síndrome de encefalopatía posterior reversible.
La hipouricemia renal hereditaria se confirma por el análisis molecular de los dos genes que codifican los transportadores de urato a nivel del túbulo renal. La hipouricemia renal tipo 1 (OMIM 220150) con pérdida de función en el gen SLC22A2 que codifica el transportador URAT1 y la hipouricemia renal tipo 2 (OMIM 612076) con mutaciones en el gen SLC2A9 que codifica el transportador GLUT9. Las formas más graves se producen en pacientes con mutaciones en el gen SLC2A9 en homocigosis. La mayoría de mutaciones se han descrito en adultos Japoneses, y sólo unos pocos casos en niños. Presentamos tres casos de niños españoles asintomáticos con hipouricemia renal confirmada genéticamente y realizamos revisión de los casos pediátricos con estudio genético, publicados en la literatura.
Hereditary renal hypouricemia is a rare autosomal recessive genetic disorder that involves an isolated defect in uric acid reabsorption at the renal tubules. Patients present with serum uric acid concentrations of less than 2mg/dl (119 micromol/L) with increased fractional excretion above 10%. Most of the patients are asymptomatic and are detected incidentally. However, complications such us nephrolithiasis, hematuria, acute renal failure exercise-induced or after dehydration for acute gastroenteritis, or posterior reversible encephalopaty syndrome (PRES) may develop.
Hereditary renal hypouricemia is confirmed by molecular genetic analysis of the two genes which codify the uric acid transport in the kidney tubules. The renal hypouricemia type 1 (OMIM 220150) is characterized by loss-of-function mutations in the SLC22A12 gene which encodes URAT 1 transporter, and the hypouricemia type 2 (OMIM 612076) is caused by defects in the SLC2A9 gene. Homozygous mutations of SLC2A9 cause the most severe forms of the disease.
Most mutations have been identified in Japanese adults, and only a few in children.
We describe three asyntomatic pediatric Spanish patients with renal hypouricemia, with genetic confirmation, and we make a revision of all of the pediatric cases with genetic study published in the literature.
La hipouricemia renal hereditaria (HRH) es un trastorno genético poco frecuente e infradiagnosticado, incluido en el grupo de enfermedades raras (ORPHA 94088). Es causado por un defecto aislado en el transporte renal del ácido úrico a nivel tubular renal, bien por disminución en su reabsorción y/o por un aumento de su secreción1,2. En niños mayores de un año y adultos, se debe sospechar ante un nivel sérico persistente de ácido úrico (AU) menor de 2mg/dl (119 micromol/l), con una excreción fraccional de AU (EFAU) mayor del 10% (normal 7,25 ± 2,98%)3–5. La HRH se confirma por el análisis molecular de los 2 genes conocidos que codifican los transportadores de AU a nivel tubular: el gen SCL22A12 y el gen SLC2A94,6. El gen SLC22A12 codifica el transportador URAT1 localizado en la membrana apical del túbulo proximal (TP). Sus mutaciones, con un patrón de herencia autosómico recesivo, son responsables de la HRH tipo 1 (OMIM 220150)7. El gen SLC2A9 codifica dos isoformas del transportador GLUT9, una larga y otra corta, localizadas respectivamente en la membrana basolateral del TP y en la apical del túbulo colector. Sus mutaciones, que siguen un patrón de herencia autosómico recesivo y en algún caso dominante, son responsables de la HRH tipo 2 (OMIM 612076)8,9.
La mayoría de los pacientes son asintomáticos, sobre todo durante la edad pediátrica, por lo que muchos no se diagnostican o se descubren de forma casual. En ocasiones, la primera manifestación puede ser hematuria o el hallazgo de hipercalciuria, pero la mayoría de las veces el diagnóstico se realiza al aparecer alguna de sus complicaciones como nefrolitiasis, daño renal agudo (DRA) inducido por el ejercicio físico intenso o menos frecuente, tras un episodio de deshidratación por gastroenteritis aguda por rotavirus10–17 o el síndrome de encefalopatía posterior reversible (SEPR) en pacientes con DRA asociado con el ejercicio18–20.
La prevalencia de la HRH es desconocida y solo existen algunos datos sobre prevalencia de hipouricemia21–24.
En la literatura se han comunicado más de 150 pacientes con HRH tipo 1, la mayoría adultos asiáticos (Japón, Corea del Sur), siendo la mutación más frecuente (2,30-2,37%) la p.W258X, y solo un escaso número se han publicado con HRH tipo 225–30.
Estudios recientes muestran que esta enfermedad no está limitada a la población asiática ya que los estudios genéticos han permitido diagnosticar pacientes europeos de diferentes grupos étnicos6,27–29,31.
Se han descrito en todo el mundo 47 mutaciones del gen SCL22A12 (36 de ellas de cambio de sentido o sin sentido) y 25 del gen SLC2A9 (15 de ellas de cambio de sentido o sin sentido), de las que 30 y 13 respectivamente, producen enfermedad32.
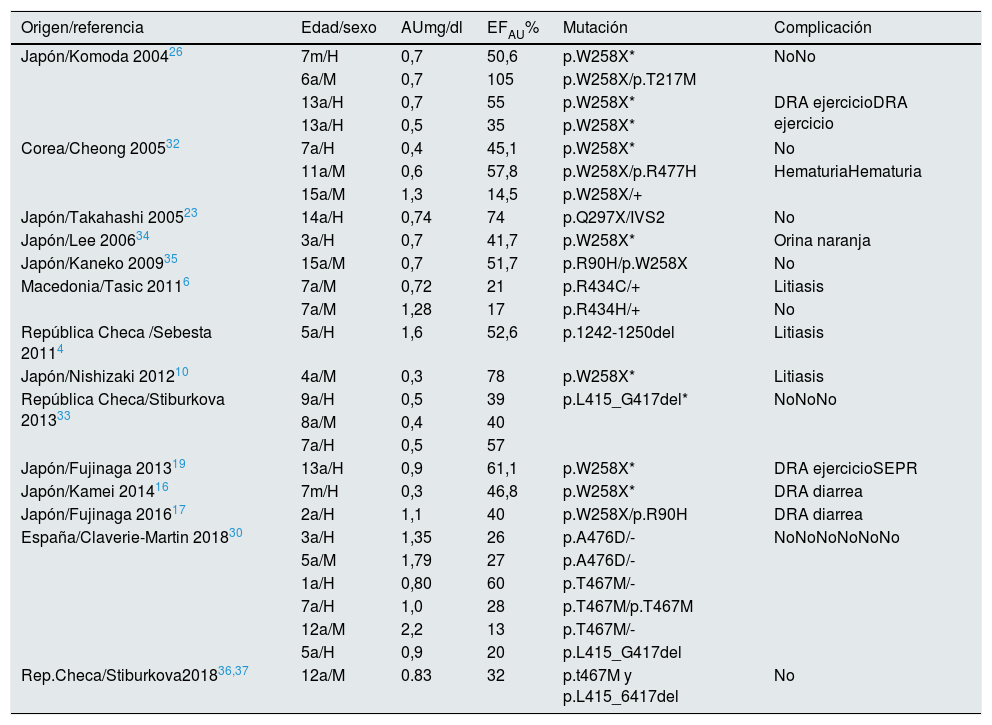
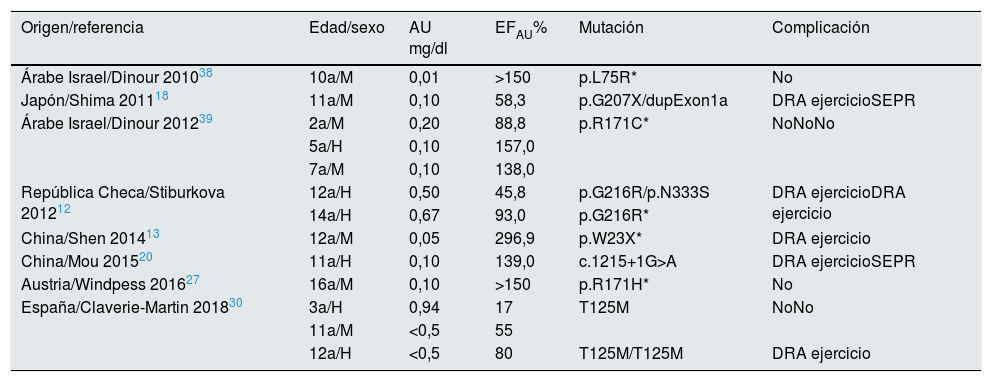
Hemos realizado una revisión bibliográfica de los casos pediátricos publicados con estudio genético. Los datos analíticos, mutaciones encontradas y manifestaciones clínicas más relevantes se recogen en las tablas 1 y 24,6,10,12,13,16–20,23,26,27,30,32–39. En total 27 niños con HRH tipo 1 y 14 con HRH tipo 2, de los que 9 casos son españoles30.
Niños con HRH tipo 1: mutaciones descritas en el gen SLC22A12
| Origen/referencia | Edad/sexo | AUmg/dl | EFAU% | Mutación | Complicación |
|---|---|---|---|---|---|
| Japón/Komoda 200426 | 7m/H | 0,7 | 50,6 | p.W258X* | NoNo |
| 6a/M | 0,7 | 105 | p.W258X/p.T217M | ||
| 13a/H | 0,7 | 55 | p.W258X* | DRA ejercicioDRA ejercicio | |
| 13a/H | 0,5 | 35 | p.W258X* | ||
| Corea/Cheong 200532 | 7a/H | 0,4 | 45,1 | p.W258X* | No |
| 11a/M | 0,6 | 57,8 | p.W258X/p.R477H | HematuriaHematuria | |
| 15a/M | 1,3 | 14,5 | p.W258X/+ | ||
| Japón/Takahashi 200523 | 14a/H | 0,74 | 74 | p.Q297X/IVS2 | No |
| Japón/Lee 200634 | 3a/H | 0,7 | 41,7 | p.W258X* | Orina naranja |
| Japón/Kaneko 200935 | 15a/M | 0,7 | 51,7 | p.R90H/p.W258X | No |
| Macedonia/Tasic 20116 | 7a/M | 0,72 | 21 | p.R434C/+ | Litiasis |
| 7a/M | 1,28 | 17 | p.R434H/+ | No | |
| República Checa /Sebesta 20114 | 5a/H | 1,6 | 52,6 | p.1242-1250del | Litiasis |
| Japón/Nishizaki 201210 | 4a/M | 0,3 | 78 | p.W258X* | Litiasis |
| República Checa/Stiburkova 201333 | 9a/H | 0,5 | 39 | p.L415_G417del* | NoNoNo |
| 8a/M | 0,4 | 40 | |||
| 7a/H | 0,5 | 57 | |||
| Japón/Fujinaga 201319 | 13a/H | 0,9 | 61,1 | p.W258X* | DRA ejercicioSEPR |
| Japón/Kamei 201416 | 7m/H | 0,3 | 46,8 | p.W258X* | DRA diarrea |
| Japón/Fujinaga 201617 | 2a/H | 1,1 | 40 | p.W258X/p.R90H | DRA diarrea |
| España/Claverie-Martin 201830 | 3a/H | 1,35 | 26 | p.A476D/- | NoNoNoNoNoNo |
| 5a/M | 1,79 | 27 | p.A476D/- | ||
| 1a/H | 0,80 | 60 | p.T467M/- | ||
| 7a/H | 1,0 | 28 | p.T467M/p.T467M | ||
| 12a/M | 2,2 | 13 | p.T467M/- | ||
| 5a/H | 0,9 | 20 | p.L415_G417del | ||
| Rep.Checa/Stiburkova201836,37 | 12a/M | 0.83 | 32 | p.t467M y p.L415_6417del | No |
a: años; AU: ácido úrico sérico; DRA: daño renal agudo; EFAU: excrección fraccional de ácido úrico; H: hombre; m: meses; M: mujer; SEPR: síndrome de encefalitis posterior reversible; *: homocigosis; +: alelo salvaje; -: no mutación en el otro alelo.
Niños con HRH tipo 2: mutaciones descritas en el gen SLC2A9
| Origen/referencia | Edad/sexo | AU mg/dl | EFAU% | Mutación | Complicación |
|---|---|---|---|---|---|
| Árabe Israel/Dinour 201038 | 10a/M | 0,01 | >150 | p.L75R* | No |
| Japón/Shima 201118 | 11a/M | 0,10 | 58,3 | p.G207X/dupExon1a | DRA ejercicioSEPR |
| Árabe Israel/Dinour 201239 | 2a/M | 0,20 | 88,8 | p.R171C* | NoNoNo |
| 5a/H | 0,10 | 157,0 | |||
| 7a/M | 0,10 | 138,0 | |||
| República Checa/Stiburkova 201212 | 12a/H | 0,50 | 45,8 | p.G216R/p.N333S | DRA ejercicioDRA ejercicio |
| 14a/H | 0,67 | 93,0 | p.G216R* | ||
| China/Shen 201413 | 12a/M | 0,05 | 296,9 | p.W23X* | DRA ejercicio |
| China/Mou 201520 | 11a/H | 0,10 | 139,0 | c.1215+1G>A | DRA ejercicioSEPR |
| Austria/Windpess 201627 | 16a/M | 0,10 | >150 | p.R171H* | No |
| España/Claverie-Martin 201830 | 3a/H | 0,94 | 17 | T125M | NoNo |
| 11a/M | <0,5 | 55 | |||
| 12a/H | <0,5 | 80 | T125M/T125M | DRA ejercicio |
a: años; AU: ácido úrico sérico; DRA: daño renal agudo; EFAU: excrección fraccional de ácido úrico; H: hombre; m: meses; M: mujer; SEPR: síndrome de encefalitis posterior reversible; *: homocigosis; +: alelo salvaje; -: no mutación en el otro alelo.
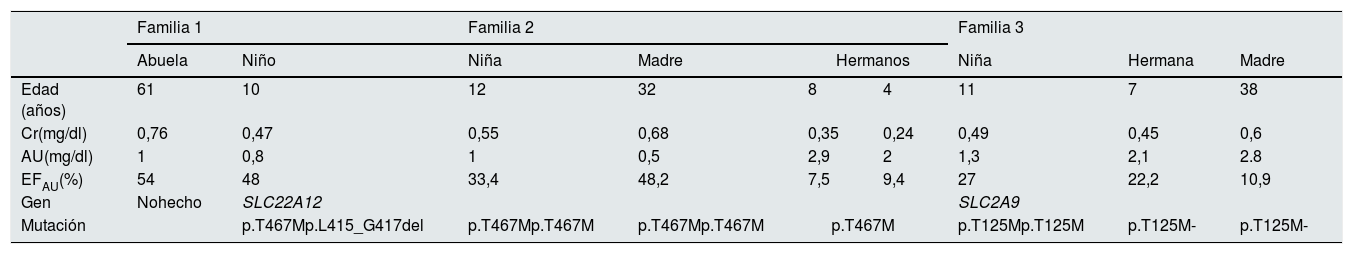
Presentamos dos niños con HRH tipo 1, de dos familias de etnia romaní no emparentadas, y un niño caucásico con HRH tipo 2 (tabla 3).
Datos clínicos y genéticos de los pacientes y sus familiares
| Familia 1 | Familia 2 | Familia 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Abuela | Niño | Niña | Madre | Hermanos | Niña | Hermana | Madre | ||
| Edad (años) | 61 | 10 | 12 | 32 | 8 | 4 | 11 | 7 | 38 |
| Cr(mg/dl) | 0,76 | 0,47 | 0,55 | 0,68 | 0,35 | 0,24 | 0,49 | 0,45 | 0,6 |
| AU(mg/dl) | 1 | 0,8 | 1 | 0,5 | 2,9 | 2 | 1,3 | 2,1 | 2.8 |
| EFAU(%) | 54 | 48 | 33,4 | 48,2 | 7,5 | 9,4 | 27 | 22,2 | 10,9 |
| Gen | Nohecho | SLC22A12 | SLC2A9 | ||||||
| Mutación | p.T467Mp.L415_G417del | p.T467Mp.T467M | p.T467Mp.T467M | p.T467M | p.T125Mp.T125M | p.T125M- | p.T125M- | ||
AU: ácido úrico sérico; Cr: creatinina plasmática; EFAU: excreción fraccional de ácido úrico
Familia 1. Niño de 10 años, sin antecedentes personales de interés; peso y talla en p50. Padres no consanguíneos, de etnia romaní, sin antecedentes nefrourológicos conocidos. Hermanos de 8 y 4 años sanos. Abuelos paternos primos hermanos. La abuela paterna refería cólicos renales. En un análisis realizado por control de tratamiento con metilfenidato en 2 ocasiones destacó un AU sérico de 0,6mg/dl. En un análisis realizado 4 años antes por anorexia, la cifra era de 0,7mg/dl, dato que pasó «desapercibido». Tras diagnosticar hipouricemia mantenida en niño asintomático con transaminasas normales, se estudió la función renal. En muestra aislada de orina no se detectó glucosuria, proteinuria, ni hipercalciuria siendo la EF AU del 48%. El equilibrio ácido-base y la ecografía renal fueron normales. Tras revisar los datos familiares, la abuela paterna tenía una hipouricemia mantenida entre 0,9-1,1mg/dl, no valorada previamente y la EFAU de 54%. Ante la sospecha de hipouricemia renal y tras consentimiento informado, se realizó estudio genético en el niño, encontrando dos mutaciones en heterocigosis en el gen SLC22A12 (p.T467M y p.L415_G417del). La abuela no permitió su estudió genético y tampoco se consiguió estudiar a los padres y hermanos porque vivían en otra comunidad.
Familia 2. Niña de 12 años, sin antecedentes de interés; peso en p97 y talla en p50. Abuelos maternos y padres, primos hermanos, de etnia romaní. Antecedentes de cólico nefrítico en la madre, abuela y tía materna. Hermanos sanos. A los 3 años en estudio de anorexia, se detectó anemia ferropénica y un AU sérico de 0,4mg/dl, tras tratamiento con hierro oral se realizó análisis de control a los 6 meses, persistiendo la hipouricemia (0,5mg/dl). En muestra aislada de orina no se detectó glucosuria, proteinuria ni hipercalciuria, y la EFAU era de 12%. Un cambio de domicilio impidió continuar con el estudio. Reapareció a los 10 años, asintomática, con AU sérico de 1mg/dl y EFAU de 33% y se completaron las exploraciones diagnósticas en ella y en su madre, que tras revisar su historia presentaba también hipouricemia persistente, AU sérico de 0,5-0,6mg/dl, no valorada, a pesar de presentar cólicos nefríticos. En ambas, el equilibrio ácido-base y la ecografía renal fueron normales. Ante la sospecha de HRH y tras consentimiento informado, se solicitó también estudio a los hermanos que tenían cifras de AU sérico normales. El estudio genético demostró que la madre y la niña son homocigotas y los 2 hermanos heterocigotos, para la mutación p.T467M del gen SLC22A12. La tía materna que tenía hipouricemia de 0,6mg/dl y la abuela no permitieron su estudio.
Familia 3. Niña de 11 años con antecedente de episodio de pielonefritis aguda; peso y talla en p50. Padres no consanguíneos, de raza caucásica, sin antecedentes nefrourológicos conocidos. Hermanas de 7 y 2 años de edad, sanas. En el análisis sanguíneo realizado durante el episodio de pielonefritis y durante su seguimiento, destacan concentraciones séricas de AU entre 1,2 y 1,8mg/dl, datos que pasaron inicialmente «desapercibidos». En el estudio de orina, no se detectó glucosuria, proteinuria ni hipercalciuria, y la EFAU fue del 27%. Ante la sospecha de HRH por hipouricemia asintomática con EFAU aumentada y tras consentimiento informado, se realiza estudio genético, que demostró una mutación en homocigosis del gen SLC2A9 (p.T125M). Se amplió estudio a familiares de primer grado, con el hallazgo de que la madre y la hermana de 7 años, son portadoras en heterocigosis de la misma mutación.
Análisis mutacional de los genes SLC22A12 y SLC2A9Utilizando muestras de sangre periférica de los pacientes y sus familiares se aisló el DNA genómico utilizando un kit comercial (Gen Elute Blood Genomic DNA kit, Sigma-Aldrich, St. Louis, MO, EE. UU.). Los exones codificantes de SLC22A12 y SLC2A9 se amplificaron mediante PCR, y se analizaron por secuenciación automática con el kit BigDye Terminator v3.1 Cycle Sequencing en el 3500 Series Genetic Analyzer (Applied Biosystems, Foster City, CA, EE. UU.). Las secuencias de DNA se compararon con sus respectivas secuencias de referencia (SLC22A12, NCBI: NG_008110.1; SLC2A9, NCBI: NG_011540.1).
ComentariosEste es el único trabajo que incluye una revisión bibliográfica de todos los casos pediátricos con HRH y la primera publicación en España de casos pediátricos confirmados genéticamente. Presentamos dos niños de etnia romaní con HRH tipo 1: un niño heterocigoto compuesto (p.T467M/p.L415_G417) y un homocigoto (p.T467M/ p.T467M), junto con dos hermanos heterocigotos para la mutación p.T467M/+. Ambas mutaciones han sido descritas previamente en niños y adultos de etnia romaní, españoles y de la República Checa28–30. La frecuencia de estas mutaciones en la cohorte romaní de España son mayores que en la República Checa (9,19 y 4,17% vs. 5,56 y 1,87%), y son las más altas del mundo, lo que sugiere una elevada incidencia de HRH en este grupo étnico, donde la endogamia y la litiasis renal son frecuentes29. Recientemente, se han descrito tres niños españoles con la mutación p.T467M, uno homocigoto y dos heterocigotos, junto con tres hermanos adultos homocigotos para esta mutación30.
La niña con HRH tipo 2, de raza caucásica, presenta la mutación p.T125M en homocigosis en el gen SLC2A9, mientras que la hermana y la madre son portadoras en heterocigosis. Esta mutación fue primero identificada en un adulto de 84 años judío-sefardí, y posteriormente en tres niños españoles, dos de ellos asintomáticos y uno que presentó DRA inducido por ejercicio físico intenso20,30.
El conocimiento de la etnia es importante en el diagnóstico y facilitar la búsqueda del tipo de mutación.
En la edad pediátrica, se debe definir hipouricemia como una concentración sérica de AU menor de 2mg/dl en mayores de un año. Esta definición es consecuencia de que entre los 2 y 12 meses de vida, debido a la «inmadurez tubular», la EFAU está aumentada (entre 27 ± 21%)4 y el nivel de AU sérico relativamente disminuido (entre 2,2-2,5mg/dl). A partir del año, la EFAU desciende a 8 ±6% y el AU sérico aumenta entre 3,5-4,5mg/dl, manteniéndose estos niveles hasta los 12 años, y a partir de esta edad las cifras son similares a las del adulto4,5.
Queremos llamar la atención sobre la importancia que tiene la hipouricemia, ya que al no tener síntomas reconocidos, este dato analítico con frecuencia pasa desapercibido o se atribuye a un error de laboratorio. Toda hipouricemia debe ser estudiada. El diagnóstico diferencial se realiza en función de la EFAU3; si esta se encuentra aumentada (mayor del 10%) es de origen tubular renal, bien en forma de tubulopatía compleja (síndrome de Fanconi primario o secundario) o de una tubulopatía aislada (HRH).
Aunque todos nuestros pacientes están asintomáticos, y se descubrieron de forma casual, se sabe que la HRH presenta una importante variabilidad clínica. Solo un 10% de los pacientes con mutaciones en el gen SLC22A12 presentan clínica, mientras que los pacientes con mutaciones en homocigosis del gen SLC2A9 son lo que pueden presentar síntomas más graves debido a su mayor EFAU (incluso mayor del 150%)8,9..
La nefrolitiasis se presenta en el 10% de los adultos con defecto en el transportador URAT1 y en el 40% de los que la alteración se encuentra en el transportador GLUT93. Esta complicación se ha comunidado en tres pacientes pediátricos con mutación en el gen SLC22A124,6,10.
La aparición brusca de DRA h o días después de realizar un ejercicio físico, como una carrera de corta distancia o tras un episodio de gastroenteritis aguda por rotavirus, debe orientar al diagnóstico de HRH. Esta complicación, se ha comunicado en cuatro niños con mutación en el gen SLC22A1216,17,19,26 y cinco en el gen SLC2A9, uno de ellos español de 12 años caucásico12,13,18,20,30. Los síntomas iniciales suelen ser dolor lumbar o abdominal junto con náuseas y fatiga muscular, que pueden confundirse con un proceso viral y demorar el diagnóstico. Ante un fallo renal agudo con una concentración sérica de AU «relativamente baja», debe plantearse la posibilidad de esta enfermedad e investigar si existen determinaciones de AU previas y repetirlas tras la normalización de la función renal, la cual en la mayoría de los casos se produce tras unos días sin que se requiera diálisis3.
Los mecanismos patogénicos propuestos de DRA asociado con el ejercicio en la HRH son:
- 1.
Aumento de la producción de AU durante el ejercicio y de su eliminación urinaria, lo que junto a una orina más concentrada (por la hipovolemia relativa a la escasa ingesta de líquidos), y ácida (por el ejercicio) facilita la precipitación y obstrucción intratubular de AU40.
- 2.
La disminución de la capacidad antioxidante, debida a la hipouricemia, posibilita que el aumento de radicales libres durante el ejercicio físico provoquen una disfunción endotelial y vasoconstricción de las arterias renales41.
- 3.
En pacientes con pérdida de función de los transportadores de urato (URAT 1 y GLUT 9), no solo está disminuida la reabsorción tubular de AU, sino que además está reducida la secreción de aniones orgánicos a la luz tubular, acumulándose y dañando las células del TP38.
El interés del diagnóstico de esta complicación es establecer medidas preventivas para evitar recidivas: limitar el ejercicio físico anaeróbico, mantener una adecuada hidratación (antes, durante y tras el ejercicio físico) con ingesta abundante de agua, tomar antioxidantes (vitamina C, carotenos), e incluso tratamiento con alopurinol37.
Se han descrito dos mecanismos por los que actuaría el alopurinol:
- 1.
Disminución de la producción de AU, con menor cantidad de AU filtrado, disminuyendo el riesgo de precipitación intratubular del AU40.
- 2.
Mejoría del daño endotelial al reducir el estrés oxidativo vascular42.
El síndrome de encefalopatía posterior reversible (SEPR) es una entidad clínico-radiológica que se caracteriza por cefalea, disminución del nivel de conciencia, convulsiones y alteraciones visuales, con imágenes en la resonancia magnética cerebral de edema cerebral, más intenso en la sustancia blanca parietooccipital. Se ha descrito en pacientes con HRH y DRA asociado al ejercicio y recientemente en pacientes pediátricos: un niño de 13 años con mutación en el gen SLC22A12 y dos de 11 años con mutación en el SLC2A918–20. Se piensa que la presencia de un aumento brusco de la presión arterial sería responsable de un edema vasogénico cerebral. El diagnóstico precoz junto con el control adecuado de las convulsiones y de la hipertensión, evitando un exceso de fluidos intravenosos, son el fundamento para prevenir la aparición de secuelas neurológicas43.
Conceptos clave1. La hipouricemia es un dato analítico que con frecuencia pasa desapercibido o se atribuye a un error de laboratorio. Toda hipouricemia por debajo de 2mg/dl debe estudiarse.
2. En niños mayores de un año, con AU <2mg/dl y EFAU >10% se debe sospechar HRH.
3. El diagnóstico de HRH se confirma por el análisis molecular de 2 genes, el gen SLC22A12 (HRH tipo1) el gen SLC2A9 (HRH tipo2).
4. La mayoría de los pacientes con HRH son asintomáticos. Las complicaciones más frecuentes son hematuria macro- o microscópica, hipercalciuria, litiasis y DRA tras ejercicio físico.
5. Para evitar las complicaciones y la recurrencia del DRA, se recomienda ingesta abundante de líquidos, antes, durante y después del ejercicio, e incluso el tratamiento con alopurinol.
El análisis genético fue financiado por PI14/00760, integrado en el Plan Nacional de I+D+I 2013-2016 y cofinanciado por el ISCIII-Subdirección General de Evaluación y Fomento de la Investigación y el Fondo Europeo de Desarrollo Regional «Una manera de hacer Europa».



