

Fabry disease (FD) is an X-linked genetic disorder characterized by alpha-galactosidase deficiency, leading to the accumulation of globotriaosylceramide. This accumulation causes multi-organ dysfunction, with renal involvement being particularly significant. Recently, the immunological relationship of this disease has been investigated, including the inactivation of enzyme therapies by antibodies and systemic inflammation. We present the case of a 15-year-old patient with FD and ANCA-associated vasculitis (AAV). A narrative review was conducted by searching PubMed with the terms “Fabry disease” AND “vasculitis” AND “glomerulonephritis,” identifying 9 relevant articles. These cases were compared with the current one, emphasizing pathophysiological aspects. 75% of patients had fever, 50% had peripheral edema, and 25% had otorhinolaryngological involvement. Pauci-immune necrosis was found in 75%. Therapeutically, all cases were treated with plasmapheresis, 75% with cyclophosphamide, and only one case required hemodialysis during follow-up. The association of FD with vasculitis is rare, with only five cases, only one with positive ANCA. The role of the immune system in FD, still not fully understood, seems to contribute significantly to pathogenesis and complications. This case highlights the need for further research on the immunological role in FD and its relationship with vasculitis and other autoimmune diseases.
La enfermedad de Fabry (EF) es un trastorno genético ligado al cromosoma X caracterizada por deficiencia de alfa-galactosidasa, que conduce a la acumulación de globotriaosilceramida. Dicha acumulación causa una disfunción multiorgánica, siendo particularmente significativo el compromiso renal. Recientemente, se ha investigado la relación inmunológica de esta enfermedad, incluyendo la inactivación de las terapias enzimáticas por parte de los anticuerpos y la inflamación sistémica. Presentamos el caso de un paciente de 15 años con EF y vasculitis asociada a ANCA (VAA). Se realizó una revisión narrativa mediante la búsqueda en PubMed de los términos «Fabry disease» Y «vasculitis» Y «glomerulonephritis», identificando 9 artículos relevantes. Dichos casos fueron comparados con el actual, destacando los aspectos patofisiológicos. El 75% de los pacientes tuvo fiebre, el 50% edema periférico y el 25% compromiso otorrinolaringológico. Se encontró necrosis pauciinmune en el 75% de los casos. Terapéuticamente, todos los casos fueron tratados con plasmaféresis, el 75% con ciclofosfamida, y solo un caso requirió hemodiálisis durante el seguimiento. La asociación de la EF con vasculitis es rara, encontrándose solo 5 casos, siendo únicamente uno positivo a ANCA. El rol del sistema inmunológico en la EF, que aún no se comprende plenamente, parece contribuir significativamente a su patogenia y complicaciones. Este caso destaca la necesidad de investigación futura sobre el rol inmunológico en la EF y su relación con vasculitis y otras enfermedades autoinmunes.
Fabry disease (FD) is an X-linked genetic disorder with an autosomal dominant inheritance pattern. The pathogenic substrate is based on the enzymatic deficiency of alpha-galactosidase, leading to systemic deposition of globotriaosylceramide, which is responsible for the multi-organ dysfunction observed in these patients.1–3 Among its clinical manifestations, renal involvement is prominent, typically presenting as albuminuria and progressive decline in glomerular filtration rate.1 Furthermore, the effect of systemic inflammation mediated by the increased expression of pro-inflammatory cytokines is being evaluated in the context of lysosomal dysfunction, which could condition the development of systemic autoimmune diseases.2–4 Specifically, the development of systemic vasculitis in the context of FD is something that, although very infrequent, is a paradigmatic fact of this mentioned immunological activation within FD.3-10
The aim of our study is to present a very unusual clinical case with the particularities that the uniqueness of our case entails, along with a narrative review of the evidence and characteristics of the cases published in the literature to provide greater evidence to the current body of this novel and interesting clinical and immunological association.
Material and methodsWe present an uncommon clinical case describing the association between ANCA-associated vasculitis (AAV), FD, and renal failure. A narrative review was conducted through a literature search in PubMed using the terms “Fabry disease” AND “vasculitis” AND “glomerulonephritis”, retrieving articles published since 1990. A total of 9 articles were collected, 4 of which corresponded to published cases of glomerulonephritis in the context of vasculitis in patients with FD. The remaining articles either did not associate acute kidney failure with ANCA-related vasculitis or were related to basic or preclinical research studies, so they were not considered for an in-depth analysis. Additionally, we have conducted an analysis of articles addressing the relationship between FD and autoimmunity, regardless of whether it is associated with acute kidney failure. This includes a discussion on the connection between FD, autoimmunity, and the potential role of enzyme replacement therapy (ERT) in these patients.
The current case is described and compared with previously published cases (Table 1), alongside a review of the most significant pathophysiological aspects linking these two rare entities. Informed consent was obtained in writing from the patient and her legal representatives for the publication of her data and images in this article.
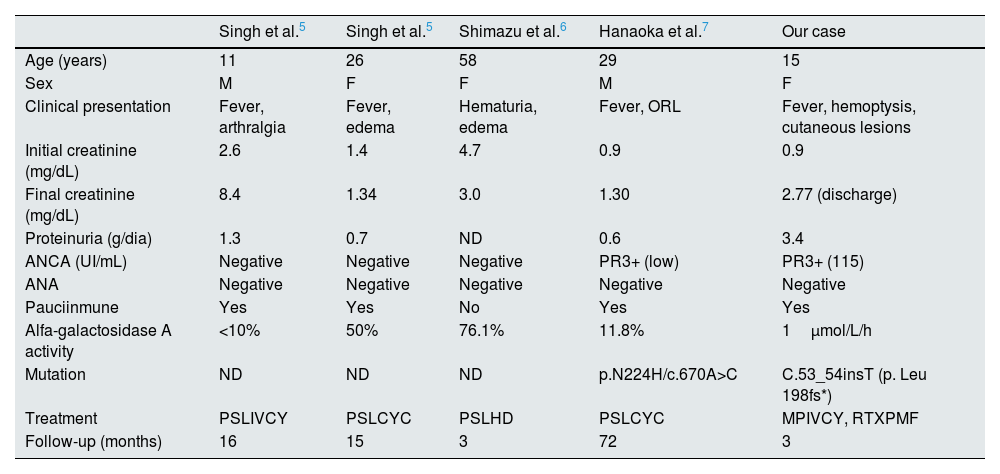
Published cases of FD associated with rapidly progressive glomerulonephritis.5–7
| Singh et al.5 | Singh et al.5 | Shimazu et al.6 | Hanaoka et al.7 | Our case | |
|---|---|---|---|---|---|
| Age (years) | 11 | 26 | 58 | 29 | 15 |
| Sex | M | F | F | M | F |
| Clinical presentation | Fever, arthralgia | Fever, edema | Hematuria, edema | Fever, ORL | Fever, hemoptysis, cutaneous lesions |
| Initial creatinine (mg/dL) | 2.6 | 1.4 | 4.7 | 0.9 | 0.9 |
| Final creatinine (mg/dL) | 8.4 | 1.34 | 3.0 | 1.30 | 2.77 (discharge) |
| Proteinuria (g/dia) | 1.3 | 0.7 | ND | 0.6 | 3.4 |
| ANCA (UI/mL) | Negative | Negative | Negative | PR3+ (low) | PR3+ (115) |
| ANA | Negative | Negative | Negative | Negative | Negative |
| Pauciinmune | Yes | Yes | No | Yes | Yes |
| Alfa-galactosidase A activity | <10% | 50% | 76.1% | 11.8% | 1μmol/L/h |
| Mutation | ND | ND | ND | p.N224H/c.670A>C | C.53_54insT (p. Leu 198fs*) |
| Treatment | PSLIVCY | PSLCYC | PSLHD | PSLCYC | MPIVCY, RTXPMF |
| Follow-up (months) | 16 | 15 | 3 | 72 | 3 |
HD: hemodyalisis; M: male; F: female; PSL: prednisolone; IVCY: intravenous cyclophosphamide; CYC: oral cyclophosphamide; MP: methylprednisolone; RTX: rituximab; PMF: plasmapheresis. ND: non-declared.
A 15-year-old female patient with a history of FD diagnosed through genetic testing revealing the mutation C.53_54insT (p. Leu 198fs*) with an insertion in heterozygosis in the X chromosome (chrX100662838 position). The basal alpha galactosidase-A activity was 1μmol/L/h (reference 2.1–14μmol/L/h) with elevated levels of Lyso-G3b on dried blood spots at diagnosis. This variant was not previously described as pathogenic in the literature. The same mutation has been detected on his father and he was also diagnosed with FD (cardiac and renal damage). She has been receiving treatment with agalsidase beta at a dosage of 1mg/kg for the past 8 months. She presented to the Emergency Department after experiencing five days of fever up to 39°C and odynophagia. Empirical antibiotic treatment with amoxicillin was initiated without response. Therefore, she returned with persistent symptoms and the appearance of petechial lesions on her lower limbs that were not raised. Upon arrival at the Emergency Department, a blood test was performed, showing leukocytosis with neutrophilia (16,540leukocytes/mm3 with 11,432neutrophils/mm3) and elevated acute phase reactants (PCR 195mg/L and procalcitonin 0.55ng/mL). Renal function at admission was slightly above the normal (creatinine 0.95mg/dL, basal values between 0.56 and 0.78mg/dL), and there was not basal proteinuria. Additionally, a chest X-ray and electrocardiogram were performed, both of which were normal. Given these findings, the decision was made to admit her to Internal Medicine for further diagnosis and treatment.
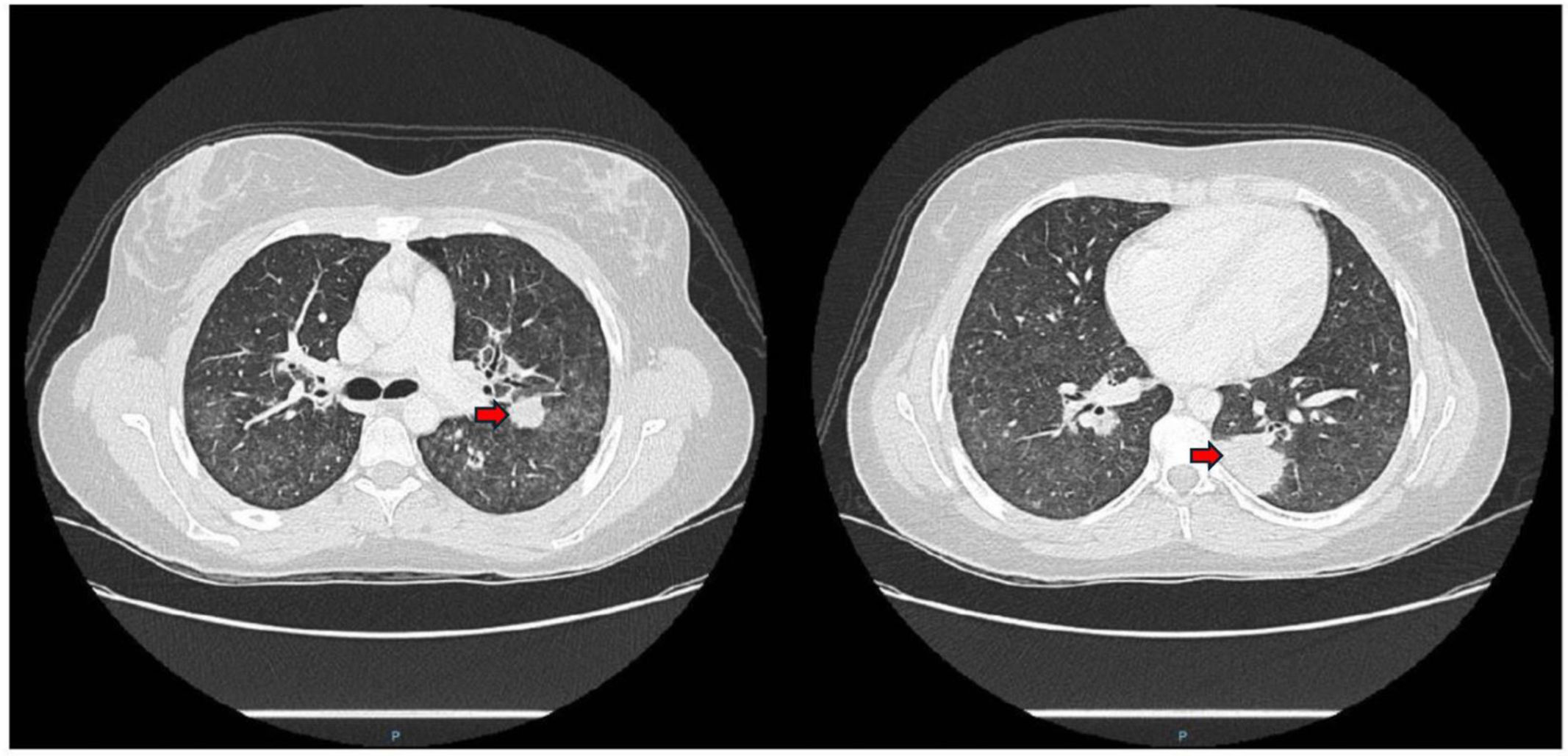
During the first 24h of hospitalization, she developed an episode of chest pain and sudden dyspnea, prompting urgent D-dimer levels to be measured (6.834ng/mL). A pulmonary artery CT angiogram revealed alveolar consolidations with the “reverse halo sign” in a peribronchovascular distribution, generalized increased density of lung parenchyma with a “tree-in-bud” pattern affecting the small airways (Fig. 1). In light of these findings, an initial analytical study was completed, including tests for autoimmunity, with negative ANA and ENA, and positive c-ANCA. The latter showed specificity for anti-proteinase-3 (PR-3) at a titer of 109UI/mL. Complement levels were normal (C3 160mg/dL and C4 29.3mg/dL), and despite normal renal function and urine analysis, a 24-h urine biochemical study was requested, revealing proteinuria levels of 3080mg/dL. Furthermore, a fiber-optic bronchoscopy was performed to better characterize the etiology of the lesions, revealing hematic crusts in both nasal vestibules and bleeding in the upper airways without signs of alveolar hemorrhage. The analysis of bronchoalveolar fluid was compatible with normality.
 are observed in both parenchyma with the reverse halo sign and a tree-in-bud pattern along with ground-glass opacities.")
Considering all clinical, imaging, and analytical data, the main diagnostic suspicion, fulfilling the classification criteria of the ACR/EULAR 2022 guidelines,5 was granulomatosis with polyangiitis (GPA), scoring a total of 10 points (5 points or more are needed to classify our patient as such), based on otorhinolaryngological involvement with hematic crusts, positive ANCA anti-PR3, and nodular pulmonary infiltrates. To rule out the possibility of treatment-induced autoimmunity, levels of antibodies against agalsidase beta were tested and found to be negative.
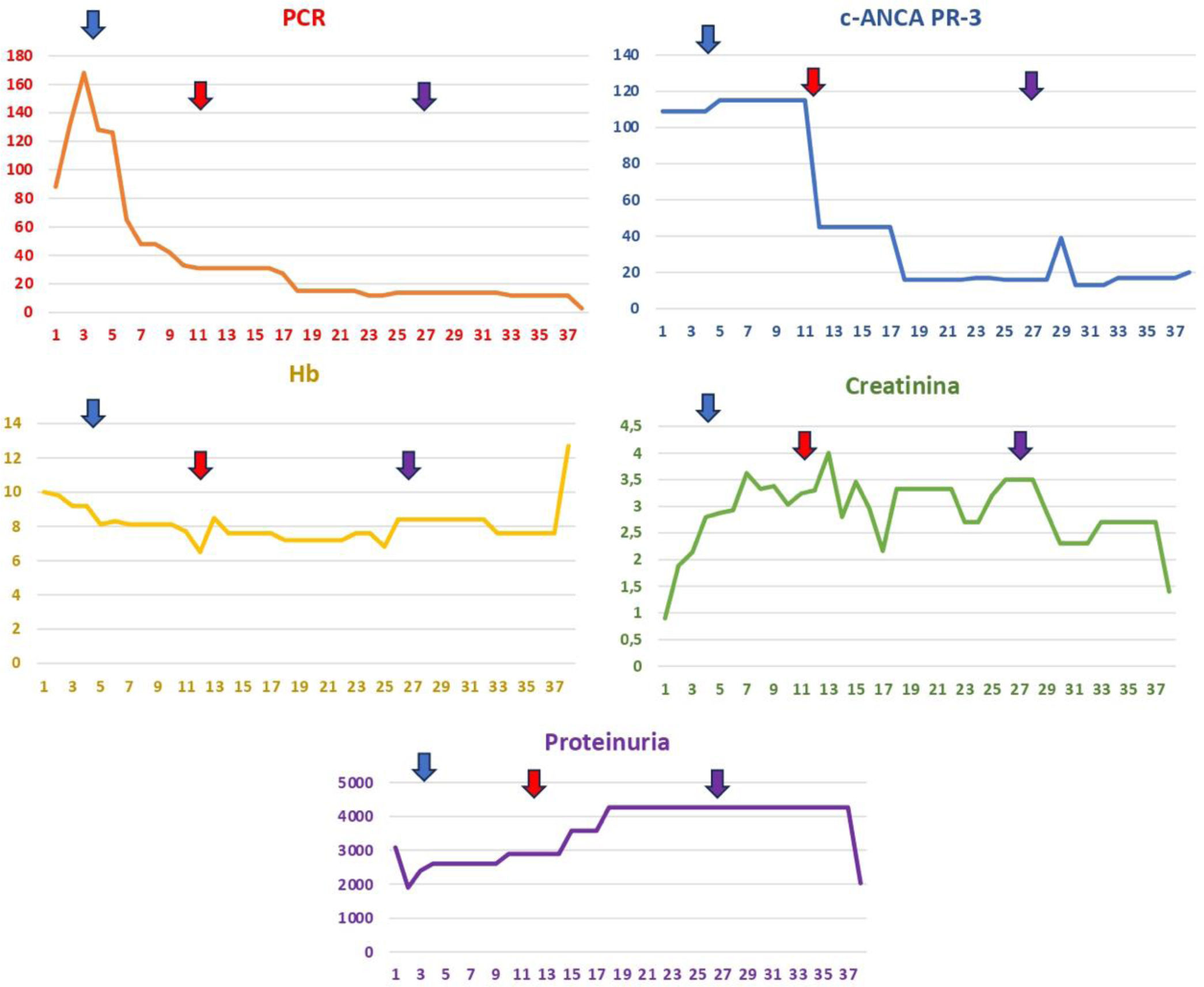
The evolution of analytical parameters is summarized in Fig. 2. Therapeutically, pulses of methylprednisolone were initiated at a dose of 250mg per day for three days (days +6, +7, and +8), followed by a dose reduction to 1mg/kg, after which there was a progressive deterioration of renal function. Due to the worsening of renal function, methylprednisolone pulses were reintroduced at a dose of 250mg per day for five days (days +12 to +17), alongside 500mg of cyclophosphamide on day +12 and a second dose of 500mg on day +27, along with rituximab at 500mg, first dose on day +12 and second dose on day +27. A renal biopsy was performed, revealing extracapillary proliferative glomerulonephritis without immune complex deposits (pauci-immune) with the formation of epithelial/fibroepithelial crescents and marked fibrinoid necrosis (Fig. 3). The pathological anatomy sample showed a degree of chronicity of 0 out of 10 points. Given that the evolution of renal function was not as expected and >90% crescents were observed, it was decided to add plasma exchange to the previous treatment scheme, with a total of seven sessions on alternate days. Progressively, renal function improved, with creatinine levels decreasing from around 4mg/dL to 2.77mg/dL before hospital discharge.
, hemoglobin (Hb), creatinine, c-ANCA PR-3, and proteinuria over several days. The blue arrow indicates the administration of boluses of methylprednisolone at a dosage of 250mg daily for three consecutive days. The red arrow denotes an escalation of treatment involving methylprednisolone at the same dosage for five consecutive days, in addition to intravenous cyclophosphamide (500mg), intravenous rituximab (500mg), and seven sessions of plasma exchange on alternate days. Finally, the purple arrow marks the second dose of intravenous cyclophosphamide (500mg) and the second dose of intravenous rituximab (500mg). These therapeutic interventions aim to control systemic inflammation and improve the patient")
The image illustrates the analytical evolution of the patient during her hospital stay, showcasing parameters such as C-reactive protein (CRP), hemoglobin (Hb), creatinine, c-ANCA PR-3, and proteinuria over several days. The blue arrow indicates the administration of boluses of methylprednisolone at a dosage of 250mg daily for three consecutive days. The red arrow denotes an escalation of treatment involving methylprednisolone at the same dosage for five consecutive days, in addition to intravenous cyclophosphamide (500mg), intravenous rituximab (500mg), and seven sessions of plasma exchange on alternate days. Finally, the purple arrow marks the second dose of intravenous cyclophosphamide (500mg) and the second dose of intravenous rituximab (500mg). These therapeutic interventions aim to control systemic inflammation and improve the patient's clinical parameters, reflecting her response to the applied treatments; CRP: C-reactive protein (mg/L); Hb: hemoglobin (g/dL); creatinine (mg/dL); c-ANCA PR-3 (IU/mL); proteinuria (protein/creatinine ratio mg/mg).
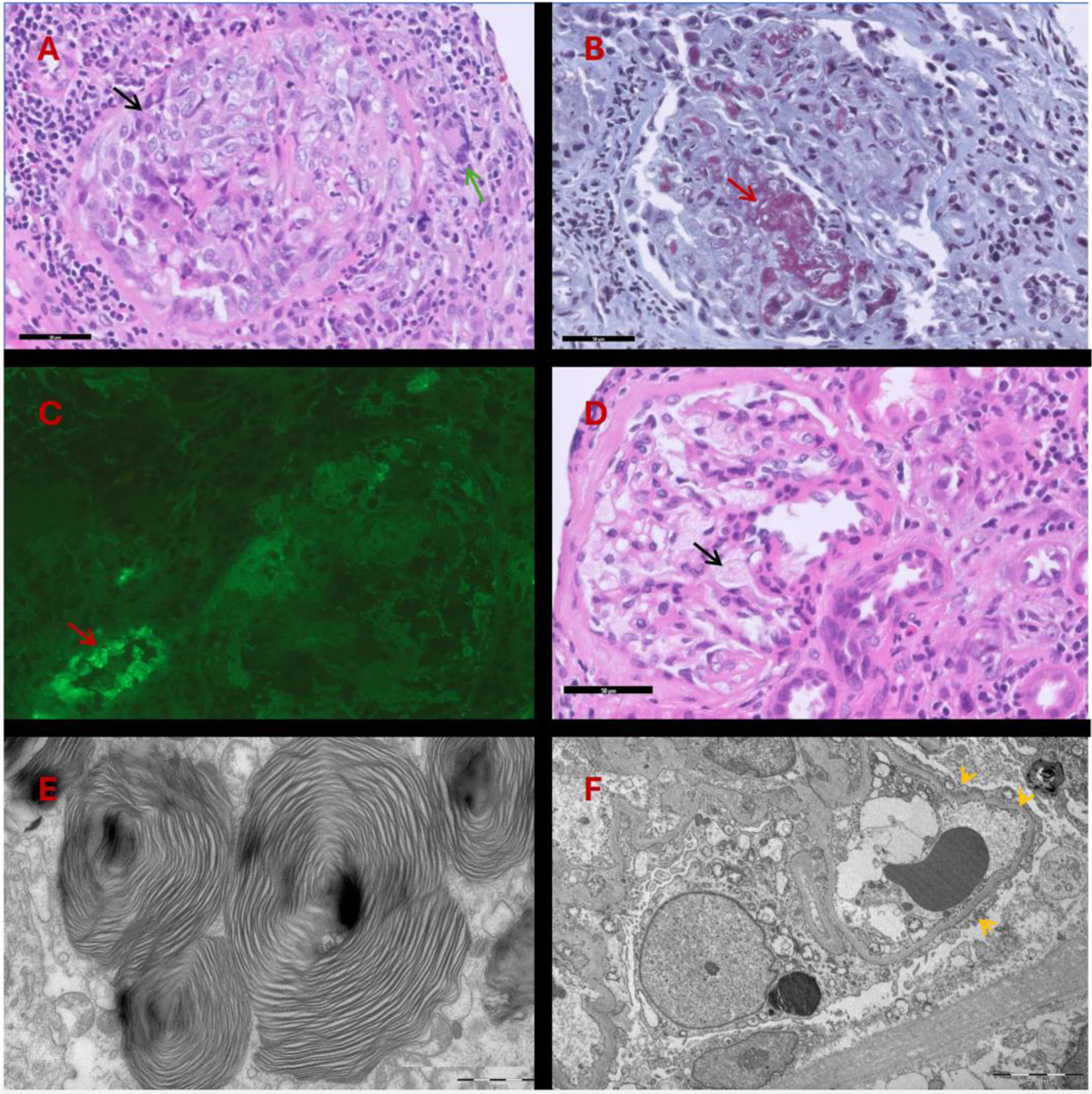
 H&E staining. Glomerulus with epithelial crescent formation (black arrow) showing rupture of Bowman")
(A) H&E staining. Glomerulus with epithelial crescent formation (black arrow) showing rupture of Bowman's capsule, chronic periglomerular inflammatory reaction with giant cells (green arrow). (B) Masson's trichrome. Marked fibrinoid necrosis (red arrow) of the glomerular capillary. (C) Immunofluorescence. C3. Absence of deposits in the glomerulus (nonspecific deposits in the arteriole wall: red arrow). (D) H&E staining. Visceral epithelial cells with foamy cytoplasm (black arrow). (E) Transmission Electron Microscopy. Intracytoplasmic laminated lipid deposits visible in podocytes, endothelial cells, and tubular epithelial cells. (F) Transmission Electron Microscopy (panoramic view): marked podocyte foot process effacement (yellow arrows).
During hospitalization, she experienced an episode of deep vein thrombosis in the left upper limb associated with the positioning of a peripheral central venous catheter without associated complications; therefore, she was anticoagulated with therapeutic doses of low molecular weight heparin. Chronic disease anemia with an iron deficiency component required transfusions during admission, along with erythropoietin and intravenous iron, all with good tolerance.
Finally, after a favorable clinical and analytical evolution, hospital discharge was decided with prednisone at a dose of 30mg per day in a tapering regimen, along with a maintenance regimen of rituximab. During subsequent follow-up in outpatient clinics, renal function improved, reaching 1.4mg/dL of creatinine, and proteinuria levels decreased to 2.035mg/mg in the protein/creatinine ratio. Hemoglobin levels also improved following the reduction of the inflammatory process, with growth factors increasing to 12.7g/dL, leaving the patient in a state of clinical and analytical remission.
DiscussionInflammation, autoimmunity and FDFD and autoimmune diseases share complex inflammatory mechanisms that contribute to both tissue damage and clinical manifestations. The accumulation of globotriaosylceramide (Gb3) and globotriaosylsphingosine (Lyso-Gb3) in lysosomes triggers an inflammatory cascade that progresses through three main phases. In the initial phase, the buildup of Gb3 affects tissues early, generating nonspecific and subclinical symptoms. In the clinical phase, chronic inflammation mediated by pathways such as NF-κB manifests as multisystemic damage, including endothelial dysfunction, oxidative stress, and progressive fibrosis. Finally, in the advanced phase, the accumulated damage leads to failure of key organs, such as the heart, kidneys, and nervous system.11–13At the molecular level, the onset of this dysregulated immune response is rooted in alterations to the innate immune response, triggering an inflammatory cascade mediated by toll-like receptors (TLRs), particularly TLR4, leading to increased production of inflammatory cytokines such as IFN-alpha and TNF-alpha. While the adaptive immune system is thought to play a less prominent role in FD-related inflammation, some hypotheses suggest that NK cells may contribute to antigen presentation to lymphocytes, acting as a bridge between innate and adaptive immunity in the autoimmune/autoinflammatory response in FD.13 A notable link identified in recent years is the complement system. Although its primary role is as an innate immune mechanism, the complement system also plays a fundamental role in adaptive immunity through processes such as opsonization, phagocytosis, and antigen presentation.13,14 We must consider that there is also an increase in autoantibodies in FD, stemming from the complex interaction between innate and adaptive immunity, with a component of autoreactivity driven by the exposure of autoantigens derived from acute and chronic tissue damage in FD, as well as the potential immunogenicity of Gb3 and Lyso-Gb3 accumulation.11–14 This chronic inflammatory process parallels autoimmune diseases, where immune dysregulation perpetuates inflammation, exacerbating tissue damage. Furthermore, inflammation in FD not only amplifies direct damage caused by Gb3 accumulation but also impairs the body's ability to resolve inflammatory processes, potentially worsening coexisting autoimmune conditions. Consequently, some authors, such as Rozenfeld et al., hypothesize that FD may be classified as an autoinflammatory disease.13,15
Several studies have explored the relationship between FD and autoimmune/autoinflammatory phenomena at both molecular and clinical levels. Fabry disease has been linked to systemic lupus erythematosus, IgA vasculitis, polyarteritis nodosa (PAN), systemic sclerosis, autoimmune thyroiditis, and classic rheumatologic conditions such as juvenile idiopathic arthritis and rheumatoid arthritis.16–23 In a case of PAN,18,19 a patient presented with acute intestinal ischemia, and histopathological analysis revealed intestinal vasculitis characterized by neutrophilic infiltrates, fibrinoid necrosis in medium-sized vessels (“pauci-immune”), and glycosphingolipid accumulation in most blood vessels. Positive staining for C3 and IgM was noted, suggesting immune complex involvement. Similarly, lesions consistent with cutaneous PAN have demonstrated C3 and IgM deposits in medium-sized vessels, with both cases exhibiting fibrolamellar structures typical of FD. These findings suggest that FD-induced vascular damage may expose endothelial antigens, promoting autoimmune responses and facilitating immune complex formation through mechanisms such as the immunogenicity of galactocerebroside or modifications of immunoglobulins by Gb3. Regarding systemic lupus erythematosus (SLE) and renal involvement,16 biopsies have shown mesangial proliferation, immunoglobulin and complement deposits, subendothelial and mesangial deposits on electron microscopy, and the characteristic cytoplasmic granules of FD. These overlapping features underscore a potential shared immunopathogenic pathway. Additionally, in IgA vasculitis associated with FD,17 six cases have been reported where aberrant glycosylation of IgA1 is hypothesized to predispose patients to both autoimmune and vasculitic processes with renal involvement, suggesting a broader role of glycosylation abnormalities in the disease's immunological context. Together, these findings highlight the intricate relationship between FD, autoimmune phenomena, and vasculitis.
Autoimmunity and ERT in FDERT and oral chaperone therapy (such as migalastat) are currently used in FD to prevent the accumulation of metabolites and tissue damage. However, it is unclear whether ERT modulates the inflammatory response. Some studies report elevated levels of proinflammatory cytokines following ERT, while others describe a reduction, leading to a lack of consensus.13,24 There are no studies yet on chaperone therapies and their interaction with the immune system. It has been established that the efficacy of ERT is linked to the development of anti-drug antibodies (ADA). Patients lacking endogenous enzyme activity are at higher risk of developing ADA, which has also been associated with the use of agalsidase beta compared to agalsidase alpha (notably, our patient was on agalsidase beta). ADA levels are correlated with decreased ERT efficacy, exacerbating systemic tissue damage caused by FD and potentially further increasing this damage.13,14,24
This hypothesis is supported by findings showing that despite a significant reduction in Lyso-Gb3 levels with ERT, there is marked complement activation, as evidenced by elevated levels of C3a and C5a, particularly in patients with nonsense mutations who developed ADA. These findings suggest that complement activation, mediated by both classical and alternative pathways, persists independently of glucolipid reduction and may contribute to multiorgan damage. Additionally, elevated levels of proinflammatory cytokines such as IL-6 and TGF-β1 have been identified, which may drive renal inflammation and fibrosis, even under ERT. These findings open the possibility of utilizing therapies targeting other molecular pathways, such as complement inhibitors, in subgroups of patients with ADA and lack of treatment response due to ADA.13 It is important to differentiate this type of hypersensitivity reaction from type 1, which is associated with infusion-related reactions that can be extremely severe, potentially leading to anaphylactic shock.13,14,24
Attempting to establish a relationship between ERT and autoimmunity on the reviewed published cases linking FD to autoimmune disorders, we observed a wide variability in treatments and dosages, ranging from 0.2mg/kg of weekly ERT to the 1mg/kg administered to our patient. Alpha-galactosidase A activity also varied significantly, from undetectable levels (<10%) to as high as 76.1%, as reported by Shimazu et al.7 Consequently, we cannot propose any hypothesis regarding the relationship between treatment or dosage and the development of autoimmune complications, nor with alpha-galactosidase A activity. In fact, in a reported case of cutaneous PAN,19 a potential link between treatment and the development of PAN was suggested but later dismissed, as the complication arose despite the patient having discontinued treatment weeks prior. Any of the published articles have measured levels of ADA. In our case, these ADA were negative. This highlights that the relationship between FD and autoimmunity extends beyond the treatment itself or the development of ADA against ERT.
Coexistence of renal disease in FD and AAVThe association of FD and vasculitis is extremely rare, with only five cases of similar characteristics found in the literature, of which only one was associated with AAV. FD and AAVs (granulomatosis with polyangiitis – GPA, eosinophilic granulomatosis with polyangiitis – EGPA, and microscopic polyangiitis – MPA) can affect the kidneys, heart, central or peripheral nervous system, and lungs, with the most significant manifestations being renal involvement.5,6 Beyond the academic classification of AAVs, the genetic component and characterization of ANCA are crucial, as it has been documented that patients with anti-MPO and anti-PR3 have different genotypes that correlate better with the specificity of the ANCA antibody than with the histological classification of vasculitis itself. Another important aspect in this clinical scenario of coexisting FD and AAV is to discern whether the organ involvement, especially at the renal level, is compatible with one entity or another, posing a significant diagnostic and therapeutic challenge.6 The renal manifestations of AAV include necrotizing glomerulonephritis without glomerular immune deposits (also referred to as “pauci-immune”), crescents in more than 50% of glomeruli, and often a rapidly progressive course. In our patient's case, not only was the vasculitic component present at the renal level, but it also manifested at the pulmonary level with findings such as pulmonary nodules with the reverse halo sign and a tree-in-bud pattern affecting the small airways (Fig. 1).6–11 Delving into the pathophysiological aspects of the condition, it has been established that vasculitis can be preceded by multiple triggers, ranging from various infectious agents to their relationship with different drugs. All of this can determine the development of immunological mechanisms that initiate a systemic inflammation mediated by autoantibodies.6 The most significant immunological mechanisms involved are molecular mimicry, autoantigen complementarity, and epitope conformation, although, specifically, vasculitis with PR-3 specificity responds to humoral and cellular components. In the same vein, the role of extracellular traps from neutrophils (NETs) containing MPO (or PR-3) and DNA in a network of histones/chromatin is being investigated. While their primary function is beneficial for combating infections by providing a crucial innate immune mechanism, excessive NET release has been associated with numerous diseases through their effector action of neutrophil-mediated damage, allowing them to circulate, present antigens to the immune system, promote hypercoagulability, and activate the alternative pathway of complement.10 NETs are also being investigated as a pathogenic determinant in FD, playing an important role in the context of ischemic cerebrovascular involvement in these patients. The neutrophil participates in innate immunity processes and plays a fundamental role in the pathophysiology of AAV, specifically in GPA. The accumulation of glycosphingolipids may increase neutrophil survival, exacerbating its pathogenic power at the level of blood vessels and favoring the process of immune-mediated vascular damage. This has been evidenced in our patient with a sluggish renal evolution despite the establishment of appropriate treatment and the occurrence of deep vein thrombosis precipitated by an endovascular catheter, favored by a pro-coagulant inflammatory environment.5,10
In the last 20 years, a total of five cases5–11 of FD associated with rapidly progressive necrotizing glomerulonephritis have been published, although only one of them met the classification criteria for GPA with positive ANCA and PR-3 specificity. The case we present would be the second in which positivity for PR-3 ANCA with a significant titer is demonstrated, unlike the one published by Hanaoka et al.,8 in which the titer is not specified but was low. Regarding proteinuria levels, our patient presented higher values than those published, reaching almost nephrotic range. Although our case has a follow-up time of less than six months and there is a possibility of improvement in glomerular filtration afterwards, all cases were left with chronic kidney disease of varying degrees. Regarding the histopathological findings, in three of the four published cases, there was pauci-immune necrotizing involvement, similar to ours. Immunofluorescence did not yield results for any antibody; however, as in the cases by Singh et al.6 and Hanaoka et al.,8 there is positivity at the level of the arteriolar walls for C3.
Regarding therapeutic aspects, all cases utilized induction with steroids and cyclophosphamide, except for Shimazu et al.7 In our case, we opted to use, in addition to the aforementioned, rituximab and plasmapheresis with an optimal response both analytically (decrease of PR-3 titers to 13UI/ml, normalization of C-reactive protein, improvement of renal function and proteinuria) and clinically (defervescence, improvement of asthenia, absence of respiratory symptoms, and disappearance of skin lesions). A biopsy of the lung tissue was not performed due to the greater prognostic and therapeutic yield of obtaining a renal sample. None of the published cases resulted in death (including the present one).
Another aspect to highlight is the difficulty of making a differential diagnosis between FD and other systemic autoimmune diseases, as there may be patients mistakenly labeled as having small-vessel vasculitis when they actually have FD, and vice versa. Our case illustrates that both entities are not incompatible, emphasizing the possibility that AAVs may be underdiagnosed when their clinical presentation is interpreted as part of FD, especially in those with isolated, rapidly progressive renal involvement leading to dialysis, where the latter is held solely responsible for the deterioration of renal function.4,6–11
Conclusions- -
FD, though rarely associated with vasculitis, exhibits evidence of immune activation involving both innate and adaptive mechanisms. This may predispose patients to autoimmune manifestations, as demonstrated by our case of FD with GPA.
- -
The coexistence of FD and AAV presents significant diagnostic challenges, particularly in discerning renal involvement attributed to FD versus AAV. Our case highlights the necessity of a comprehensive diagnostic approach, including renal biopsy and autoantibody testing, to guide appropriate management.
- -
Chronic inflammation in FD, driven by the accumulation of Gb3 and Lyso-Gb3, mirrors autoimmune mechanisms. This reinforces the hypothesis that FD may be considered an autoinflammatory condition, especially when systemic autoimmune manifestations like vasculitis are present.
- -
ERT in FD can elicit varied immune responses, including the development of ADA in some patients. In our case, while ADA was negative, persistent inflammatory markers and complement activation were noted, indicating the complexity of immune modulation in FD.
- -
The successful treatment of our patient involved high-dose methylprednisolone, cyclophosphamide, rituximab, and plasmapheresis. This underscores the need for aggressive and combined therapeutic strategies in cases with overlapping autoimmune and lysosomal storage disorders.
- -
The overlapping features of FD and vasculitis can lead to underdiagnosis of AAV in FD patients, particularly in those with rapidly progressive renal dysfunction. Our case demonstrates that these entities can coexist.
No funding was received.
Conflict of interestsNo conflicts of interest to declare.



