

La enfermedad de Castleman es un trastorno linfoproliferativo poco común, de etiología incierta. Existen 3 patrones histopatológicos con manifestaciones clínicas diversas, desde adenopatías localizadas y curso clínico benigno hasta manifestaciones sistémicas de peor pronóstico y alto riesgo de evolucionar hasta procesos malignos.
Presentamos a continuación los casos de enfermedad de Castleman con afectación renal atendidos en el Servicio de Nefrología del Hospital General Universitario de Ciudad Real, entre 2017 y 2024. Describimos la evolución clínica y el manejo terapéutico de cada uno de ellos.
Castleman's disease is a rare lymphoproliferative disorder of uncertain etiology. There are 3 histopathological patterns with different clinical manifestations, from localized adenopathies and benign clinical course to systemic manifestations of worse prognosis and high risk of developing into malignant processes.
The following are cases of Castleman's disease with renal involvement treated in the Nephrology Service of the General University Hospital of Ciudad Real, between 2017 and 2024. We describe the clinical evolution and therapeutic management of each of them.
La enfermedad de Castleman (EC), o hiperplasia linfonodular angiofolicular, es un trastorno linfoproliferativo de etiología no clara, descrita por primera ver por Benjamín Castleman en 19561.
Afecta con igual frecuencia a mujeres y hombres y suele ser más frecuente en adultos jóvenes.
Desde el punto de vista clínico, puede presentarse como una entidad localizada o unicéntrica (ECU), con aparición de uno o más ganglios linfáticos agrandados en una sola región del cuerpo y curso más indolente, o bien como una afectación múlticéntrica (ECM), con linfadenopatías generalizadas y un comportamiento más agresivo con afectación sistémica (fiebre, anemia, pérdida de peso, hepatoesplenomegalia, aumento de la velocidad de eritrosedimentación e hipergammaglobulinemia). La ECM se subclasifica, además en ECM asociada al VHH8, generalmente en pacientes inmunocomprometidos, y la ECM idiopática (ECMi), que engloba a otras 4entidades diferentes: EMCi asociada a POEMS (polineuropatía, organomegalia, endocrinopatía, trastorno monoclonal de células plasmáticas y cambios en la piel), síndrome EMCi-TAFRO (trombocitopenia, anasarca, mielofibrosis, disfunción renal y organomegalia), EMCi-LPI (linfadenopatía plasmocítica idiopática) y la EMCi-NOS o no específica2,3.
El diagnóstico de confirmación es histopatológico, con 3variantes posibles: la hialinovascular, la más frecuente (80-90%) que suele corresponder a las formas localizadas de la enfermedad; la variante plasmocelular (10%), asociada generalmente a la variante multicéntrica y con mayor frecuencia relacionada con procesos malignos, y la variante mixta (2%)4.
En relación con el tratamiento, parece haber consenso en cuanto al manejo quirúrgico de las variantes unicéntricas. No obstante, no existe hoy en día consenso en cuanto al tratamiento óptimo para la variedad multicéntrica5.
Presentamos a continuación una serie de 4casos estudiados en nuestro centro entre los años 2017 y 2023: 3 de ellos mostraron variedad hialinovascular, 2 eran multicéntricos idiopáticos, con adenopatías de predominio retroperitoneal y el otro era unicéntrico de localización cervical. El cuarto caso fue de tipo multicéntrico VHH8 positivo con histología plasmocelular. Todos ellos asociaron enfermedad renal de base, la mayoría de etiología autoinmune.
Casos reportadosCaso 1Varón de 53 años, hipertenso de larga evolución y mala adherencia terapéutica, que acudió a Urgencias por crisis hipertensiva (TA 244/144mmHg). A la exploración, destacaban edemas bimaleolares, con resto anodino. En la analítica, se objetivó anemia en rango transfusional (Hb 6,9mg/dl) sin datos de hemólisis (frotis sin esquistocitos, LDH 228 UI/L, reticulocitos 0,79%, haptoglobina 2,8mg/dl; con coagulación normal, plaquetas 219.000 mcL). Se objetivó deterioro importante de la función renal (Cr 3,8mg/dl) con proteinuria subnefrótica (cociente proteína/creatinina 0,82), sin hematuria; el sedimento no procedía. Revisando estudios previos, sin datos desde 7 años antes, se vio que entonces tenía función renal normal, sin alteraciones urinarias. Se realizó estudio de HTA secundaria, con resultado negativo o normal. Además, se solicitó protocolo glomerular con anticuerpos antinucleares (ANA), anticuerpos anticitoplasma de neutrófilos (ANCA), crioglobulinas y anticuerpos antirreceptor de fosfolipasa A2 (anti-PLA2R) negativos. Las inmunoglobulinas con IgM eran de 38 UI/ml; el resto, en rango. No había pico monoclonal (relación kapppa/lambda en suero 1,17) y complemento C3 y C4 también normal. Proteinograma sin alteraciones. Se solicitaron, además, serologías (VHC, VHB, VIH, sífilis), sangre oculta en heces y marcadores tumorales; también negativos. Se realizó biopsia renal, con diagnóstico definitivo de ERC en contexto de HTA maligna/acelerada. Durante el ingreso, se detectó una adenopatía inguinal palpable de nueva aparición, por lo que se solicitó TAC-body, que fue informada como hepatoesplenomegalia y adenopatías subcentimétricas supra- e infradiafragmáticas, llamativas en número, y con captación en PET-TAC, todo ello sugestivo de síndrome linfoproliferativo crónico. En la biopsia de la adenopatía inguinal, se describieron cambios compatibles con enfermedad de Castleman hipervascular, VHH8 y VEB negativo.
Se inició tratamiento con corticoterapia a dosis de 1mg/kg día y siltuximab. Evolucionó de manera tórpida, con aparición de pancitopenia y empeoramiento de la función renal (Cr 5,6mg/dl) con aumento de proteinuria hasta rango nefrótico (6,5 por cociente proteína/creatinina), síndrome nefrótico clínico y analítico. Como otras alteraciones asociadas aparecieron hipotiroidismo grave e hipergammaglobulinemia policlonal, no presente previamente. En este contexto, se produjo insuficiencia respiratoria aguda y disminución del nivel de consciencia, con necesidad de traslado a UCI, donde finalmente se produjo el fallecimiento por complicaciones cardiacas (tormenta arrítmica) a los 2meses del inicio de la enfermedad hematológica.
Caso 2Varón de 66 años, que acudió al Servicio de Urgencias por astenia, pérdida de peso y disnea progresiva de varias semanas de evolución. En alguna ocasión, febrícula. En la radiografía de tórax, se apreciaba una imagen nodular redondeada en lóbulo superior del pulmón derecho. Se amplió el estudio con TAC y se confirmó la presencia de nódulo subpleural derecho bien delimitado de 9×7mm, sin actividad metabólica en PET-TAC. Además, se describieronn adenopatías supra- e infradiafragmáticas (laterocervicales mediastínicas, axilares, retroperitoneales periaórticas e interaortocava e inguinales bilaterales, las mayores de 12mm). Sin organomegalias. Durante el ingreso, el paciente desarrolló un síndrome nefrótico clínico y bioquímico (cociente proteína/Cr 6,5g) con microhematuria (sedimento 3-5 hematíes/campo) y función renal normal (Cr 0,85mg/dl). Revisando históricos, se vio que la función renal previa era normal, sin alteraciones urinarias. En el estudio inmunológico, anticardiolipina IgG (ACA) de 15 U/ml, ANCA positivo (no estudio por IFI), ANA positivo a título bajo (1/80), con anti-ADN y anticuerpos nucleares extraíbles (ENA) negativos y consumo del complemento (C3 de 57mg/dl y C4 de 10mg/dl). Además, había anemia en rango transfusional (Hb 7,2mg/dl), sin datos de hemólisis, trombocitopenia leve (plaquetas 138.000 mcL), aumento de VSG a 41 e hipergammaglobulinemia policlonal (IgG 1.875mg/dl, IgM 589mg/dl, IgA 387mg/dl). Se realizó entonces biopsia renal, informada como nefropatía membranosa con proliferación endocapilar focal y depósitos de C1q, muy sugestiva de etiología secundaria. PLA 2R negativos en biopsia, C4d positivo. Se amplió el estudio analítico con serologías (VHC, VHB, VIH, sífilis) y marcadores tumorales negativos. Biopsia de médula ósea normal y punción de nódulo pulmonar solitario sin datos de malignidad. Se realizó, finalmente, biopsia de adenopatía inguinal, en la que se describieron cambios compatibles con enfermedad de Castleman plasmocelular VHH8+(VHH8 negativo en biopsia renal).
Dado el proceso hematológico de base con afectación renal, se decidió, de forma consensuada con el Servicio de Medicina Interna, iniciar tratamiento con corticoterapia (prednisona 1mg/kg día) y rituximab (375mg por 8 dosis en total), con buena evolución cínica y analítica y consiguiendo la remisión completa a los 3 meses de tratamiento. En la actualidad, tras 4 años desde el diagnóstico y sin tratamiento inmunosupresor de mantenimiento, el paciente se mantiene asintomático, con función renal conservada y adenopatías estables.
Caso 3Mujer de 28 años que consultó por molestias en el cuello en contexto de masa cervical izquierda, indolora y de crecimiento lento. Refería también sudoración nocturna sin otra sintomatología, sequedad bucal, no ocular y dolor habitual de espalda. A la exploración, además, se apreciaron lesiones cutáneas café con leche. Se realizó TAC body con la que se confirmó la presencia de adenopatías laterocervicales bilaterales, la mayor de 1,5cm y submaxilar, en cola de parótida izquierda con actividad metabólica en PET-TAC. Sin organomegalias. Se realizó biopsia de la adenopatía cervical izquierda, objetivando cambios compatibles con enfermedad de Castleman hialinovascular VHH8 y EVB-EBER negativo. Durante el seguimiento por Medicina Interna, hizo aparición un rash malar ocasional y dolor articular habitual (manos y rodillas). Ante la sospecha de posible enfermedad autoinmune asociada (LES o enfermedad de Sjögren) nos consultaron para ampliar el estudio. En la analítica destacaban: anemia leve (Hb 10,6mg/dl) y leucocitopenia (38.000 mcL) con linfopenia leve. La función renal se mantenía normal, con hematíes y proteínas en orina (sedimento con 5-10 hematíes/campo y cociente proteínas/creatinina 0,38). Revisando históricos, ya en analíticas previas presentaba proteinuria y hematuria similares desde al menos 2 años antes. En el estudio inmunológico, ANA positivo (título1/640) con anti- ADN y ENA negativos; anticoagulante lúpico positivo a títulos altos (IgM 70, IgG 200); ACA positivo (18 U/ml) e hipocomplementemia (C3 de 78mg/dl y C4 de 6mg/dl); VSG normal.
Se inició tratamiento con corticoides a dosis bajas (prednisona 0,5mg/kg al día), siltuximab y ácido acetil salicílico por el SAF analítico. Tuvo una mala evolución clínica y radiológica y precisó ingreso en el Servicio de Nefrología por síndrome nefrótico florido (proteinuria de hasta 6 g, con hipoalbuminemia [2,8g/dl] y edemas generalizados). La función renal era normal.
En este contexto, se inició tratamiento con bolos de corticoides intravenosos (500mg por 3 dosis) junto con dosis oral a 1mg/kg al día y se programó biopsia renal, con hallazgos anatomopatológicos compatibles con nefropatía lúpica de tipo iv y alto índice de actividad (11/24). Se decidió en este punto iniciar tratamiento con rituximab (375mg por 8 dosis) para abordaje conjunto de la nefropatía lúpica y la enfermedad de Castleman y se consiguió la remisión completa de ambas enfermedades. En la evolución posterior, está estable desde el punto de vista hematológico y en remisión completa desde el punto de vista renal. Sigue un tratamiento de mantenimiento con prednisona (10mg/día) e hidroxicloroquina (200mg/día). Se detectó un pequeño brote del lupus, a los 2años del diagnóstico, con proteinuria de 1,5 gramos y mínimo deterioro reversible de la función renal (Cr 1,45mg/dl) de probable etiología prerrenal. En el sedimento: microhematuria con proteinuria por cociente de 1,49. ANA positivos (título 1/320), sin haber llegado a negativizarlo en ningún momento, con anti-ADN negativo y anticoagulante lúpico positivo (IgM 40, IgG 140). También se detectó hipocomplementemia (C3 de 70mg/dl; C4 de 6,5mg/dl). En ese momento se asoció micofenolato (500mg cada 12h), que se aumentó hasta la dosis de 1.500mg/día. Desde el punto de vista renal hubo de nuevo remisión de la enfermedad (función renal normal, con microhematuria persistente y mínima proteinuria de 0,45g). Sin embargo, persistía la clínica sistémica con artralgias y astenia, por lo que se consensuó con el Servicio de Reumatología asociar tratamiento con belimumab, con buena evolución posterior, ya desde los 6 meses de tratamiento. Actualmente, desde el punto de vista nefrológico: proteinuria residual en torno a 0,3-0,7 y función renal normal, sin adenopatías y con mejoría de la clínica sistémica. La paciente continúa con prednisona a dosis bajas (5mg/día), micofenolato (200mg cada 12h) y belimumab (10mg/kg mensual), aunque con mala adherencia terapéutica.
Caso 4Mujer de 45 años que consultó por nódulo mamario de crecimiento lento. En el estudio de imagen se describió lesión mamaria de aspecto benigno, pero, de manera incidental, se describieron también adenopatías axilares bilaterales, sin lesiones en otra zona ni organomegalias. Sin sintomatología sistémica en ese momento. Se realizó biopsia de uno de los ganglios axilares, con diagnóstico de enfermedad de Castleman hipervascular, VHH8 y VEB negativo. En el estudio complementario, la analítica con función renal era normal, sin alteraciones urinarias. Como resultado del protocolo inmunológico, ANA positivos a título bajo (1/160), con anti-ADN positivo, ENA negativos e hipergammaglobulinemia policlonal (IgG 1715mg/dl, IgM 679mg/dl, IgA 407mg/dl). Sin consumo del complemento y ANCA, ACA y anticoagulante lúpico negativos. La biopsia de médula ósea, sin datos de infiltración. En la entrevista dirigida, refirió lesiones en la piel del escote en alguna ocasión, con rash malar, aftas orales y artralgias generalizadas, que ahora no presenta. Se indicó tratamiento con hidroxicloroquina (200mg/día) y seguimiento. A los 2años del diagnóstico de la EC, empezó con síndrome nefrótico clínico y bioquímico con proteinuria de 8 g, edemas bimaleolares e hipoalbuminemia de 2,3mg/dl y microhematuria de 5 hematíes/campo en sedimento. La función renal era normal. En el análisis inmunológico, persistía la positividad de ANA, ahora a título de 1/640 con anti-ADN positivo y anti-ENA positivos (SS-A:++, Ro 52:++, SS-B:+, nucleosomas+, histonas+, AMA M2:+++). Presentaba hipergammaglobulinemia policlonal persistente y consumo del complemento (C3 de 48mg/dl y C4 de 7mg/dl). Durante este tiempo, las adenopatías permanecieron estables. Se decidió realizar entonces biopsia renal, con resultado anatomopatológico de nefropatía lúpica de tipo iv. En este contexto, se mantuvo el tratamiento con antipalúdicos (hidroxicloroquina a 200mg/día) y se inició tratamiento con corticoides intravenosos (3 bolos de 500mg) justo a dosis oral de 1mg/kg al día en pauta descendente y micofenolato (250mg cada 12h), consiguiendo buena respuesta clínica y analítica. Actualmente se mantiene en remisión completa, con función renal conservada y adenopatías estables. Mantiene tratamiento con prednisona de 2,5mg a días alternos con micofenolato en pauta descendente de 250mg al día y antipalúdico. El Servicio de Reumatología planteó la posibilidad de asociar belimumab antes de la retirada completa de micofenolato.
DiscusiónEstudiamos en nuestro servicio a 4pacientes diagnosticados de EC entre los años 2017 y 2024.
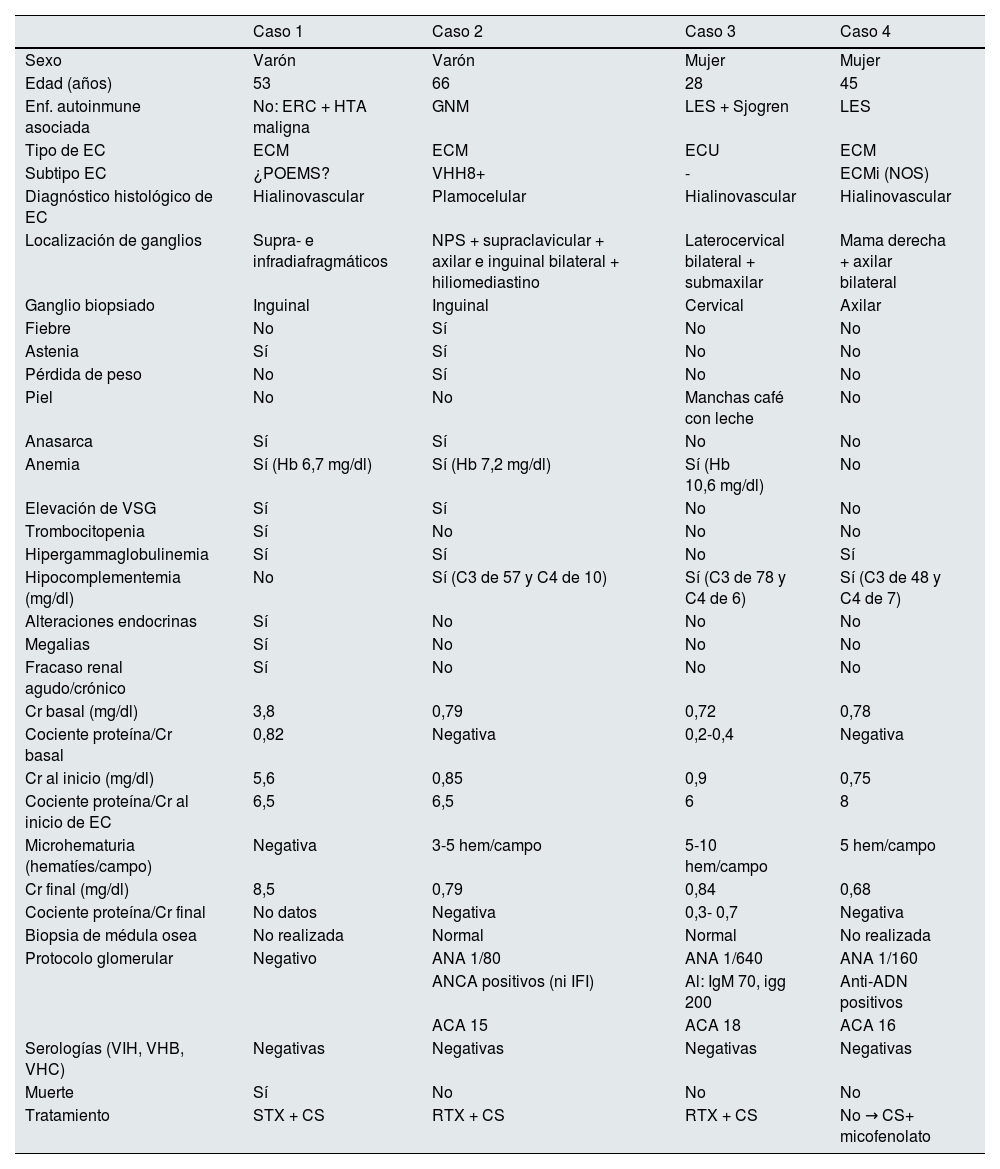
La enfermedad se presentó con igual frecuencia en hombres que en mujeres, a diferencia de lo que se reporta en la bibliografía médica1. La edad media de presentación fue de 48 años y la localización más frecuente para la presentación de las adenopatías fue la retroperitoneal. Solo en el caso de ECU se identificaron en la zona cervical. La tabla 1 recoge los datos epidemiológicos, características clínicas y resultados analíticos de todos los pacientes.
Datos epidemiológicos, características clínicas y resultados analíticos de los pacientes con enfermedad de Castleman
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | |
|---|---|---|---|---|
| Sexo | Varón | Varón | Mujer | Mujer |
| Edad (años) | 53 | 66 | 28 | 45 |
| Enf. autoinmune asociada | No: ERC + HTA maligna | GNM | LES + Sjogren | LES |
| Tipo de EC | ECM | ECM | ECU | ECM |
| Subtipo EC | ¿POEMS? | VHH8+ | - | ECMi (NOS) |
| Diagnóstico histológico de EC | Hialinovascular | Plamocelular | Hialinovascular | Hialinovascular |
| Localización de ganglios | Supra- e infradiafragmáticos | NPS + supraclavicular + axilar e inguinal bilateral + hiliomediastino | Laterocervical bilateral + submaxilar | Mama derecha + axilar bilateral |
| Ganglio biopsiado | Inguinal | Inguinal | Cervical | Axilar |
| Fiebre | No | Sí | No | No |
| Astenia | Sí | Sí | No | No |
| Pérdida de peso | No | Sí | No | No |
| Piel | No | No | Manchas café con leche | No |
| Anasarca | Sí | Sí | No | No |
| Anemia | Sí (Hb 6,7 mg/dl) | Sí (Hb 7,2 mg/dl) | Sí (Hb 10,6 mg/dl) | No |
| Elevación de VSG | Sí | Sí | No | No |
| Trombocitopenia | Sí | No | No | No |
| Hipergammaglobulinemia | Sí | Sí | No | Sí |
| Hipocomplementemia (mg/dl) | No | Sí (C3 de 57 y C4 de 10) | Sí (C3 de 78 y C4 de 6) | Sí (C3 de 48 y C4 de 7) |
| Alteraciones endocrinas | Sí | No | No | No |
| Megalias | Sí | No | No | No |
| Fracaso renal agudo/crónico | Sí | No | No | No |
| Cr basal (mg/dl) | 3,8 | 0,79 | 0,72 | 0,78 |
| Cociente proteína/Cr basal | 0,82 | Negativa | 0,2-0,4 | Negativa |
| Cr al inicio (mg/dl) | 5,6 | 0,85 | 0,9 | 0,75 |
| Cociente proteína/Cr al inicio de EC | 6,5 | 6,5 | 6 | 8 |
| Microhematuria (hematíes/campo) | Negativa | 3-5 hem/campo | 5-10 hem/campo | 5 hem/campo |
| Cr final (mg/dl) | 8,5 | 0,79 | 0,84 | 0,68 |
| Cociente proteína/Cr final | No datos | Negativa | 0,3- 0,7 | Negativa |
| Biopsia de médula osea | No realizada | Normal | Normal | No realizada |
| Protocolo glomerular | Negativo | ANA 1/80 | ANA 1/640 | ANA 1/160 |
| ANCA positivos (ni IFI) | Al: IgM 70, igg 200 | Anti-ADN positivos | ||
| ACA 15 | ACA 18 | ACA 16 | ||
| Serologías (VIH, VHB, VHC) | Negativas | Negativas | Negativas | Negativas |
| Muerte | Sí | No | No | No |
| Tratamiento | STX + CS | RTX + CS | RTX + CS | No → CS+ micofenolato |
ERC: enfermedad renal crónica; HTA: hipertensión arterial; GNM: glomerulonefritis membranosa; LES: lupus eritematoso sistémico; EC: enfermedad de Castleman; ECM: enfermedad de Castleman multicéntrica; ECU: enfermedad de Castelman unicéntrica; POEMS: polineuropatía, organomegalia, endocrinopatía, trastorno monoclonal de células plasmáticas y cambios en la piel; VHH8: herpes virus humano 8; ECMi (NOS): enfermedad de Castleman idiopática (no específica); NPS: nódulo pulmonar solitario; VSG: velocidad de sedimentación glomerular; Cr: creatinina; ANA: anticuerpos antinucleares; ANCA: anticuerpos anticitoplasma de neutrófilos; ADN: ácido desoxirribonucleico; ACA: anticuerpos anticardiolipina; VIH: virus de la inmunodeficiencia humana; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; STX: siltuximab; RTX: rituximab; CS: corticoides.
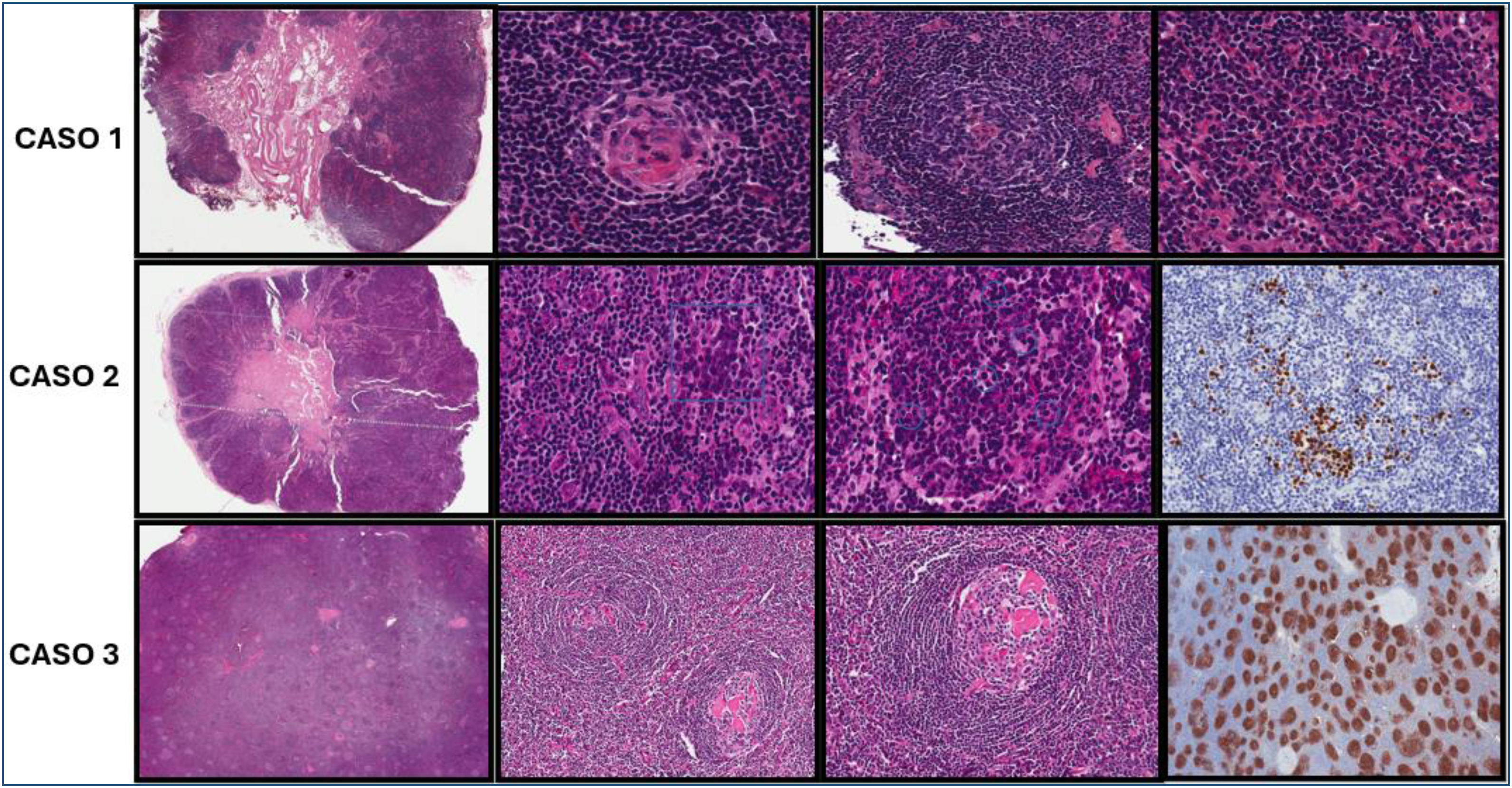
Aunque, según la bibliografía, la forma de presentación más habitual es la localizada2,5,6, en nuestro caso, 3de los 4pacientes fueron diagnosticados de ECM, 2idiopáticas y uno asociada al VHH8. La variante más frecuente fue la hialinovascular, en el 75% de los casos, y solo un caso, el multicéntrico asociado a VHH8, se correspondió a la variante plasmocelular. Según la bibliografía, la EC asociada a VHH8 se relaciona con infección por VIH en más del 40% de los casos7. En nuestro paciente, las serologías resultaron negativas. La figura 1 recoge la descripción anatomopatológica de los ganglios linfáticos.
 Panorámica de parte del ganglio linfático con medular central y cortical de patrón folicular con sinusoides dilatados. B) A mayor aumento, folículos linfoides hipoplásicos-atróficos, en los centros germinales vasos de pared hialinizada y rodeados por células linfoides del manto concéntricas («disposición en capas de cebolla»). C) En zonas interfoliculares se observa proliferación de células plasmocitoides de aspecto maduro. D) No hay imágenes de técnica IHQ, pero el resultado fue negatividad para HHV8 y EBV-EBER, así como expresión politípica de cadenas ligeras. Expresión positiva de CD30 limitada a blastos foliculares. Caso 2: A) Panorámica. Biopsia escisional de ganglio linfático con medular central y corteza ganglionar con borramiento de la arquitectura habitual del ganglio. B) Presencia de agrupaciones de blastos (recuadro). C) Marcada linfoplasmocitosis (plasmáticas marcadas con círculos). D) La IHQ muestra positividad en los blastos para HHV-8. Caso 3: A) Panorámica. Biopsia escisional de ganglio linfático con patrón folicular. B y C) A mayor aumento, folículos linfoides hipoplásico-atróficos, en los centros germinales vasos de pared hialinizada y rodeados por células linfoides del manto de forma concéntrica («disposición en capas de cebolla»). D) La IHQ muestra marcada expresión de CD23 y CD21 en centros germinales. Caso 4: Sin hay imágenes.")
Descripción anatomopatológica de ganglios linfáticos. Caso 1: A) Panorámica de parte del ganglio linfático con medular central y cortical de patrón folicular con sinusoides dilatados. B) A mayor aumento, folículos linfoides hipoplásicos-atróficos, en los centros germinales vasos de pared hialinizada y rodeados por células linfoides del manto concéntricas («disposición en capas de cebolla»). C) En zonas interfoliculares se observa proliferación de células plasmocitoides de aspecto maduro. D) No hay imágenes de técnica IHQ, pero el resultado fue negatividad para HHV8 y EBV-EBER, así como expresión politípica de cadenas ligeras. Expresión positiva de CD30 limitada a blastos foliculares. Caso 2: A) Panorámica. Biopsia escisional de ganglio linfático con medular central y corteza ganglionar con borramiento de la arquitectura habitual del ganglio. B) Presencia de agrupaciones de blastos (recuadro). C) Marcada linfoplasmocitosis (plasmáticas marcadas con círculos). D) La IHQ muestra positividad en los blastos para HHV-8. Caso 3: A) Panorámica. Biopsia escisional de ganglio linfático con patrón folicular. B y C) A mayor aumento, folículos linfoides hipoplásico-atróficos, en los centros germinales vasos de pared hialinizada y rodeados por células linfoides del manto de forma concéntrica («disposición en capas de cebolla»). D) La IHQ muestra marcada expresión de CD23 y CD21 en centros germinales. Caso 4: Sin hay imágenes.
En cuanto a la clínica, aunque según los estudios publicados la afectación multicéntrica suele presentar un comportamiento más agresivo con afectación sistémica8, en nuestra experiencia solo el tipo multicéntrico VHH8 positivo con variante plasmocelular se manifestó desde el inicio, con fiebre, anemia, astenia y pérdida de peso. Estas alteraciones parecen asociarse al aumento de secreción de IL-6 y, con frecuencia, puede complicarse con el desarrollo de amiloidosis, infecciones y enfermedades malignas, particularmente el sarcoma de Kaposi o el linfoma5,7. En el resto de los pacientes, el motivo de consulta fueron las adenopatías palpables o su hallazgo incidental en el estudio por otras causas. Podríamos plantearnos aquí que la agresividad del cuadro, más que por la clasificación anatómica, podría venir determinada por la presentación histológica y corresponder a los casos más graves, con la variante plasmocelular. Esta hipótesis ya se defiende en algunos trabajos5,7,10.
A pesar de que en ninguno de los casos de ECMi idiopáticos incluidos en el estudio se realizó subclasificación según las entidades presentes en la bibliografía (POEMS, TAFIRO, LPI, NOS), observamos que uno de nuestros pacientes, además de las manifestaciones sistémicas comentadas, presentó organomegalia, trombocitopenia y alteraciones endocrinas graves, lo que nos hizo sospechar a posteriori de la variante POEMS. Este fue el único caso de EC con afectación renal previa al diagnóstico y sin enfermedad autoinmune de base asociada. En todos los demás pacientes (casos 2, 3 y 4), encontramos un sustrato inmunológico, como el LES y GNM secundaria, diagnosticados mediante biopsia renal. La duda que nos planteamos en este punto es si nos encontramos ante manifestaciones autoinmunes de la EC o bien se trata de procesos autoinmunes cuya histología ganglionar se asemeja a la de la ECMi. Los 2casos con diagnóstico de LES (casos 3 y 4) son los que nos generan mayor incertidumbres, ya que, según el consenso de expertos para el diagnóstico integrado de la EC multicéntrica idiopática, entre el 15% y el 30% de los pacientes con lupus muestran una histopatología ganglionar similar a la de la ECMi9. No encontramos en la bibliografía trabajos que profundicen en este aspecto, aparte de la mención de enfermedades como el LES, la artritis reumatoide, el síndrome linfoproliferativo autoinmune y la enfermedad de Still como posibles criterios de exclusión para EC9,11. En nuestra serie de casos, el diagnóstico de EC fue previo al de LES, con una brecha temporal de meses incluso de años hasta el inicio de la enfermedad renal, por lo que se consideraron como entidades coexistentes, aunque con un sustrato inmunológico relacionado a efectos de tratamiento.
Del mismo modo, haciendo una revisión de la afectación renal de la EC, son muy pocos los artículos que realizan una descripción detallada en este sentido, de ahí lo interesante de nuestro trabajo. La mayoría de los estudios revisados describen de forma somera una manifestación clínica marcada por el fracaso renal agudo con aumento de la creatinina plasmática y síndrome nefrótico asociado, pero no aportan más datos en cuanto a valores analíticos y evolución cronológica2-4,9. En nuestra serie de casos, sí que hemos recogido estos aspectos. Como puede comprobarse en la tabla 1, solo el paciente del caso 1 presentó deterioro de la función renal previa al diagnóstico de la EC, aunque su etiología no se consideró relacionada con la enfermedad hematológica en sí. El resto de los casos, y a diferencia de lo que se contempla en la bibliografía, conservaron la función renal durante todo el proceso agudo. Sí que desarrollaron todos un síndrome nefrótico clínico y bioquímico, con una proteinuria marcada por encima de los 6 g, y se objetivaron alteraciones en el sedimento urinario en forma de microhematuria, algo no descrito hasta el momento. En cuanto a la autoinmunidad, los ANA estuvieron presentes en todos los pacientes, con diferente titulación, aunque su carácter inespecífico nos impide establecer conclusiones al respecto. En todos estos casos, hubo consumo del complemento, algo que ya se contempla en la EC sin afectación renal9,11. Sí que nos llamó la atención la presencia de ACA en la mayoría de los pacientes.
Revisando la bibliografía, no encontramos hoy en día ninguna mención que relacione estos autoanticuerpos con la EC, por lo que quizás sería interesante tenerlo en cuenta para futuros estudios.
Finalmente, cabe destacar los hallazgos anatomopatológicos confirmados mediante biopsia de riñón realizada a cada uno de nuestros pacientes. Dado que la afectación renal en la mayoría de los casos es muy poco frecuente, tampoco encontramos grandes estudios con un patrón claro establecido. Revisando publicaciones de casos clínicos aislados y pequeñas series de casos, parece que las 2lesiones más frecuentemente encontradas desde el punto de vista histopatológico en pacientes con EC y afectación renal fueron de microangiopatía similar a la trombótica (MAT-like) y amiloidosis secundaria12-14. Sin embargo, como puede observarse en la figura 2, la descripción anatomopatológica en nuestra muestra difiere de lo publicado hasta la fecha. En la mayoría de los casos, se describe un engrosamiento de la membrana basal, con imágenes de «apolillado» y una expansión mesangial, con proliferación endocapilar a expensas de células mononucleares y algunos neutrófilos. En el compartimento intersticial, se describe una leve fibrosis, con mínima necrosis tubular y sin infiltrado inflamatorio, o con mínima presencia de mononucleares. Tampoco se objetivó en la mayoría de los casos daño vascular alguno. El caso 1 sí presentó daño vascular, con hiperplasia intimal asociada a HTA maligna, pero en ningún momento se planteó la relación con la EC. En todos ellos, la tinción con rojo Congo fue negativa y la inmunofluorescencia directa estuvo en consonancia con el diagnóstico renal emitido, como puede verse en las imágenes.
Por último, en cuanto al tratamiento de la forma localizada, existe en la bibliografía el criterio uniforme de que debe ser tratada quirúrgicamente4,10,15. Sin embargo, en nuestro caso, en el único paciente catalogado como variante unicéntrica (caso 3), se optó por el manejo médico y el seguimiento.
Respecto al tratamiento de las variantes multicéntricas, no parece haber consenso. Se han obtenido resultados variables con quimioterapia sola, en combinación con esteroides y también con radioterapia16,14,17. En nuestro caso, el paciente número 1 fue tratado con siltuximab, quizá porque la afectación renal se atribuyó a una etiología no autoinmune desde el principio. En los otros 2pacientes con variantes multicéntricas, se prefirió el uso de rituximab para abordaje conjunto de ambos procesos. La mayoría de ellos recibieron además tratamiento con corticoides desde el inicio.
Hasta la fecha, los 3 pacientes que continúan vivos se encuentran asintomáticos, incluido el de la variante plasmocelular, y sin actividad renal de la enfermedad autoinmune. Debido a la naturaleza potencialmente maligna de la forma multicéntrica o plasmocelular, los pacientes deben ser vigilados y evaluados de forma periódica, sin que se haya descrito hasta la fecha el tiempo de seguimiento necesario para considerar la curación. No puede descartarse la aparición a largo plazo de complicaciones como el sarcoma de Kaposi o el linfoma no hodgkiniano.
ConclusiónLa EC es un trastorno linfoproliferativo benigno poco común, que se presenta, en los ganglios, como una hiperplasia folicular linfoide.
Entre sus manifestaciones clínicas, la afectación renal es muy poco frecuente; aparece en la mayoría de los casos como síndrome nefrótico clínico y bioquímico con o sin deterioro de la función renal.
No se conoce una autoinmunidad específica asociada a esta entidad. Nuestros resultados indican que los anticuerpos anticardiolipinas podrían estar presentes cuando hay procesos autoinmunes asociados, aunque se necesitarían estudios más amplios para establecer una relación directa. Tampoco encontramos un patrón histológico constante en la biopsia renal. El engrosamiento de la membrana basal, con expansión mesangial y proliferación endocapilar, son los hallazgos más repetidos en nuestro estudio. No objetivamos daño vascular ni positividad para rojo Congo.
Se hace evidente la necesidad de más estudios, así como compartir y analizar cada uno de los casos diagnosticados en cada centro, para entender mejor la patogenia, la fisiopatología y la evolución natural de esta enfermedad, ya que solo así mejorarán sus opciones diagnósticas y terapéuticas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.



