





Atypical hemolytic-uremic syndrome (aHUS) is a rare, life-threatening complement-mediated thrombotic microangiopathy.1 Approximately half of cases have mutations in complement proteins but only 12% have 2 or more mutations.2 Eculizumab is nowadays considered first-line therapy for aHUS.3
A 33-year-old female with unremarkable past medical history presented with a 3-day history of decreased urine output. Physical examination showed hypertension (160/90mmHg) and lower limbs edema. Investigations revealed an acute thrombotic microangiopathy (ATM), hematuria and nephrotic proteinuria (Table 1). Daily plasmapheresis (PMP) was started immediately.
Laboratorial results on admission and additional studies to establish the cause of the thrombotic microangiopathy.
| On admission | ||
|---|---|---|
| Variable | Result | Normal range |
| Hemoglobin (g/dL) | 11.1 | 12–16 |
| White-cell count (×103/mm3) | 7.6 | 4–10 |
| Platelet count (×103/mm3) | 36 | 150–400 |
| Peripheral blood smeara | 10 schistocytes | <2 schistocytes |
| Haptoglobin (g/L) | <0.08 | 0.3–2 |
| Direct coombs tests | Negative | Negative |
| Urea (mg/dL) | 114 | 15–38 |
| Creatinine (mg/dL) | 2.9 | 0.52–1.04 |
| Sodium (mmol/L) | 135 | 137–145 |
| Potassium (mmol/L) | 3.6 | 3.5–5.1 |
| Lactate dehydrogenase (U/L) | 2827 | 313–618 |
| c-Reactive protein (mg/dL) | 0.6 | <1.0 |
| URINALYSIS | ||
| Protein (mg/dL) | 1000 | |
| Leukocytes (/HPF) | 3 | <5/c |
| Red blood cells (/HPF) | 28 | <2/c |
| Urinary protein to creatinine ratio (mg/mg) | 14 | <0.15 |
| Additional studies | ||
|---|---|---|
| Variable | Result | Normal range |
| C3 (g/L) | 0.46 | 0.9–1.8 |
| C4 (g/L) | 0.02 | 0.1–0.4 |
| CH50 (U/mL) | 13 | 23–46 |
| C1q (mg/L) | 39 | 118–244 |
| C1q Inhibitor (mg/L) | 197 | 180/320 |
| C2 (mg/L) | 7.2 | 14–25 |
| ADAMTS13 activity (%) | 7% | 40–130 |
| Anti-ADAMTS13 antibody (U/mL) | Negative | <15 |
| Serum cryoglobulins | Positive | |
| Auto-immune antibodiesb, Pregnancy test, SPE, Tumoral markersc, HIV, HBV, HCV | Negative/Normal | |
| Blood, urine and stool cultures | Negative | |
| Upper endoscopy, colonoscopy, cervical, thoracic and abdominopelvic CT | Irrelevant changes | |
Peripheral blood smear was done to evaluate presence of erythrocyte fragmentation and is reported as schistocytes count in a microscopic field at 100× magnification.
Antinuclear antibodies, extractable nuclear antigens, anti-neutrophil cytoplasmic antibodies, glomerular basement membrane, rheumatoid factor, cyclic citrullinated peptide, anticardiolipin/anti-β2-glycoprotein, Lupus anticoagulant.
Carcinoembryonic antigen, carbohydrate antigen 19-9, α-fetoprotein, cancer antigen 125, neuron-specific enolase, NSE.
Abbreviations: ADAMTS13; a disintegrin and metalloproteinase with a thrombospondin type 1 motif; member 13. CT; computerized tomography. HBV; hepatitis B virus. HCV; hepatitis C virus. HIV; human immunodeficiency virus. SPE; serum protein electrophoresis.
Investigations for secondary causes of ATM revealed a low ADAMTS 13 activity, decreased complement C4, C3, C1q and C2 levels and positive serum cryoglobulins (Table 1). Considering the hypothesis of an autoimmune disorder, we started 3 daily pulses of 1000mg methylprednisolone followed by oral prednisolone.
PMP was stopped on the eighteenth day of admission (D18) due to normal platelet count during 3 consecutive days (Fig. 1). On the D19 a renal biopsy was made. It showed a “thrombotic microangiopathy with acute tubular necrosis” and “deposition of IgM, C3 and C1q in the capillary wall”. Due to increased hemolytic activity, PMP was resumed on the D21. On the D27, rituximab was started to enhance immunosuppression. PMP was stopped on the D35 based on the absence of schistocytosis, hemoglobin stability and lactate dehydrogenase (LDH) normalization.
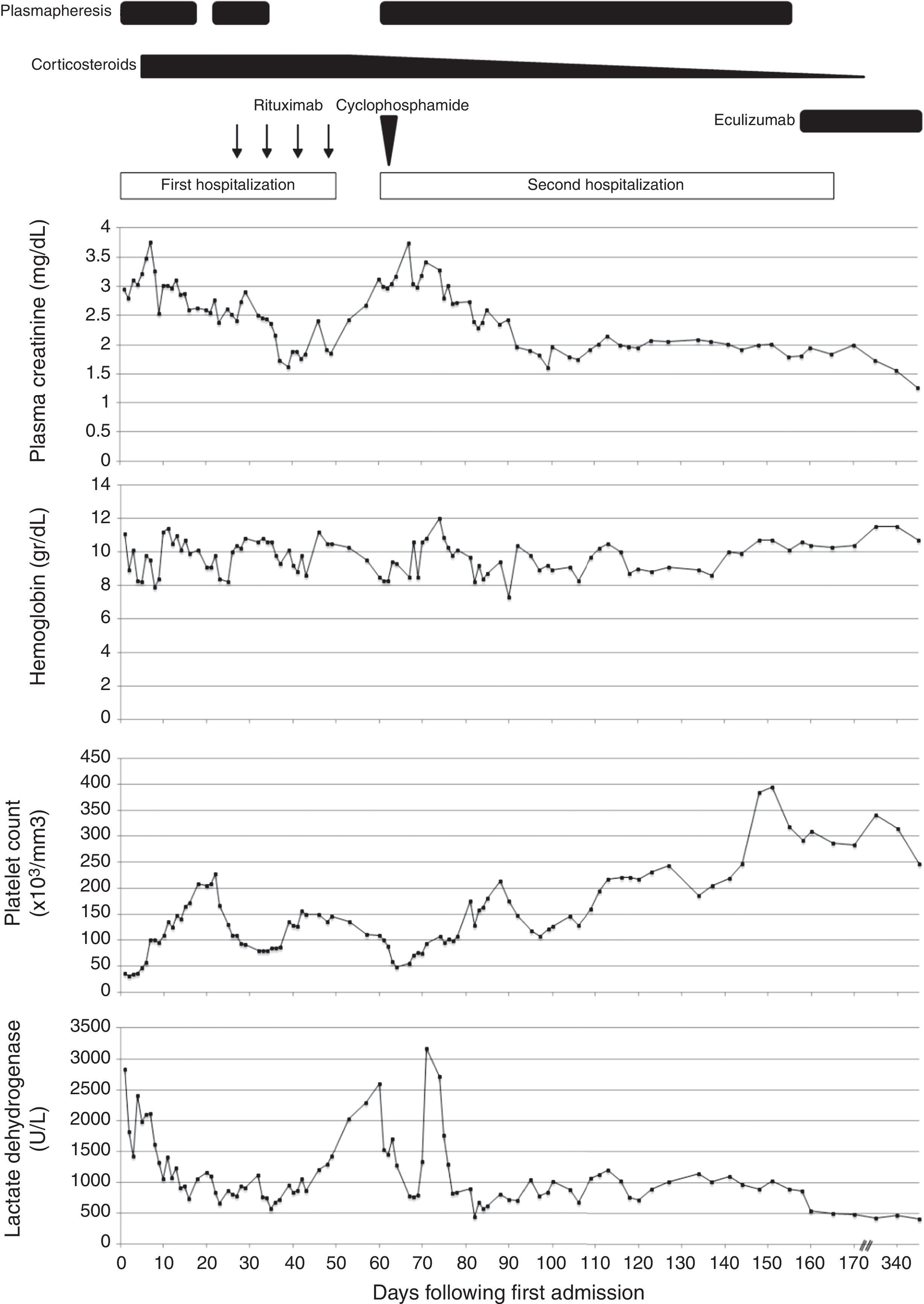
Clinical course: laboratory data and treatment. Corticosteroid therapy was started with intravenous administration of methylprednisolone (1000mg/day for 3 days) followed by oral prednisolone and an initial dosage of 60mg/day. This dosage was maintained until the 53rd day after admission, with posterior tapering. Rituximab (arrows) was administered at a dose of 600mg (375mg/m2) on the 27th, 34th, 41st and 48th days after admission. Cyclophosphamide (arrowhead) was administered in a single dose of 1000mg on the 61st day after admission (2nd day after readmission). Eculizumab was administered at a dose of 900mg for a week for 4 weeks, started on the 158th day after admission, followed by a dosage of 1200mg 1 week later an then a maintenance dose of 1200mg every 2 weeks. This dosage is still being continued. We performed a total of 79 plasma exchange sessions. The last three laboratorial test results were done on the 270th, 340th and 466th days following first admission.
On the D50, LDH was high but hemoglobin and platelet count were stable. A state of compensated hemolysis was assumed and the patient was discharged home.
Ten days later she was readmitted due to increased hemolytic activity (Fig. 1). PMP was resumed and 1000mg cyclophosphamide was given. At that time ADAMTS13 and complement C4 levels were normal but complement C3 levels remained low.
A genetic screening for mutations in complement regulatory proteins was made. Two mutations were found in factor I (C. 452 A>G, pASN 151 Ser) and C3 (C193 A>C, pLys 65 Gin) proteins. The process of eculizumab acquisition was lengthy, wherefore this therapy was started only on the 98th day after readmission. At that moment we were performing one PMP session/week, LDH remained high (859U/L) and moderate renal dysfunction (creatinine 2.08mg/dL and urea 60mg/dL) and nephrotic proteinuria persisted (5.2g/24h). Eculizumab was administered at a dose of 900mg per week for 4 weeks followed by subsequent doses of 1200mg every 2 weeks since the 5th week. Complete hematologic remission was attained 2 days after eculizumab initiation. The patient was discharged home on the 105th day after readmission. Four weeks later, proteinuria was only 0.6g/24h.
Currently, 10 months after first infusion, the patient remains under biweekly administration of eculizumab. Hematologic remission persists and there is a significant recover of renal function (urea 43mg/dL, creatinine 1.27mg/dL).
Complement factor I (CFI) and C3 mutations account for 10% and 4% of the overall aHUS-associated mutations, respectively.3 C3 mutations are among the ones with the poorest prognosis: 75% risk of death or end stage renal failure (ESRF) at 3–5 years follow up and 50% risk of recurrence.3 CFI mutations have a 50–60% risk of death and ESRF at 3–5 years follow up and 10–30% risk of recurrence.3
Both mutations present in our patient were previously reported.4,5 The p.Asn151Ser mutation causes a quantitative deficiency of factor I4 and the p.Lys65Gin mutation weakens the affinity of C3b to complement factor H.5
Despite the growing importance of eculizumab,6,7 PMP remains the mainstay of treatment while waiting for the immunoglobulin.3 Nonetheless, its benefit depends on the underlying genetic defect – only 25% and 57% of those with CFI and C3 mutations, respectively, achieve remission.8 In our patient, PMP was critical to prevent further progression of renal failure and the development of other systemic involvement. However, as described by Loirat,2 it was unable to achieve complete and sustained remission.
In our case, the confounding presence of low ADAMTS13, C4, C2 and C1q levels, the presence of cryoglobulins and the existence of immune deposits in the glomerular capillary wall led to the misdiagnosis of a thrombotic thrombocytopenic purpura secondary to an autoimmune disease. It explains the preference for glucocorticoids, rituximab and cyclophosphamide in relation to eculizumab.9
Eculizumab is nowadays considered first-line therapy for aHUS.3 It is a humanized monoclonal immunoglobulin IgG that targets C5 and blocks the uncontrolled generation of the cytotoxic membrane attack complex.10 When we started Eculizumab, our patient was still PMP dependent despite the 79 PMP sessions. Contrariwise, complete hematologic remission was attained two days after first infusion and a remarkable recover of renal function and reduction of proteinuria was evident. These findings reinforce the benefits of eculizumab in aHUS, even in cases of severe renal involvement.
In conclusion, our case highlights the diagnostic challenge of aHUS and emphasizes the notion that isolated PMP is no longer the best therapeutic option in aHUS, in which cases a prompt switch to eculizumab is mandatory.
The authors declare that they have no conflicts of interest related to the contents of this article.



