




El gen UMOD codifica la proteína uromodulina, la cual tiene una función crucial en el funcionamiento renal. Las alteraciones genéticas que afectan a su correcta función se relacionan principalmente con nefropatía tubulointersticial autosómica dominante (ADTKD), insuficiencia renal progresiva e hiperuricemia, entre otros fenotipos clínicos variables. En la población gallega existen mutaciones recurrentes en este gen, este estudio tiene como objetivo caracterizar fenotípicamente las variantes recurrentes para mejorar el pronóstico y las estrategias de manejo de los pacientes afectados.
MetodologíaSe realizó un estudio de cohorte retrospectivo con 37 pacientes de 15 familias portadoras de variantes recurrentes en UMOD (p.C255Y y p.Q316P, del transcrito NM_001008389.3) en una población gallega, caracterizada por su alta conservación genética. Se recopilaron datos clínicos, incluyendo función renal, hiperuricemia, hipertensión y presencia de quistes renales. Los análisis genómicos se realizaron mediante secuenciación masiva (NGS) y Sanger, clasificando las variantes según las guías del ACMG. Las comparaciones estadísticas se llevaron a cabo mediante pruebas de Mann-Whitney, Chi-cuadrado y Fisher, con corrección de Benjamini-Hochberg para pruebas múltiples.
ResultadosLa cohorte incluyó 28 portadores de p.C255Y y 9 de p.Q316P. Ambas variantes afectan dominios altamente conservados con baja tolerancia a los cambios de aminoácidos, lo que altera la función de la proteína y tiene efectos clínicos en los pacientes. Se observó hiperuricemia en el 76% de los portadores de p.C255Y y en el 50% de los portadores de p.Q316P, mientras que solo se asoció la primera variante con episodios de gota. Los quistes renales y la hipertensión fueron identificados en aproximadamente la mitad de los pacientes, independientemente del tipo de variante. Las curvas de Kaplan-Meier sugirieron una progresión más temprana a hiperuricemia y a enfermedad renal crónica avanzada (ERCA) en portadores de p.C255Y, aunque sin alcanzar significación estadística.
ConclusionesLas mutaciones recurrentes de UMOD en una cohorte gallega revelaron características clínicas compartidas, como hiperuricemia y progresión a ERCA, con una variabilidad fenotípica influenciada por la edad y los modificadores genéticos adicionales. Estos hallazgos subrayan el valor pronóstico de las correlaciones genotipo-fenotipo y la necesidad de un manejo clínico individualizado en los pacientes con ADTKD.
The UMOD gene encodes the uromodulin protein, which plays a crucial role in renal function. Genetic alterations affecting its correct function are mainly related to Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD), progressive renal failure and hyperuricaemia, among other variable clinical phenotypes. In the Galician population there are recurrent mutations in this gene, this study aims to phenotypically characterise the recurrent variants to improve the prognosis and management strategies of affected patients.
MethodsA retrospective cohort study was conducted with 37 patients from 15 families carrying recurrent variants in UMOD (p.C255Y and p.Q316P, from transcript NM_001008389.3) in a Galician population characterised by high genetic conservation. Clinical data were collected, including renal function, hyperuricemia, hypertension and presence of renal cysts. Genomic analyses were performed by NGS and Sanger sequencing, variant classification were conducted according to ACMG guidelines. Statistical comparisons were performed using Mann-Whitney, Chi-square and Fisher tests, with Benjamini-Hochberg correction for multiple testing.
ResultsThe cohort included 28 carriers of p.C255Y and 9 of p.Q316P. Both variants affect highly conserved domains with low tolerance to amino acid changes, which alters protein function and has clinical effects in patients. Hyperuricaemia was observed in 76% of p.C255Y carriers and 50% of p.Q316P carriers, while only the first variant was associated with episodes of gout. Renal cysts and hypertension were identified in about half of the patients, independently of variant type. Kaplan-Meier curves suggested an earlier progression to hyperuricaemia and advanced chronic kidney disease (CKD) in p.C255Y carriers, although without reaching statistical significance.
ConclusionsRecurrent UMOD mutations in a Galician cohort revealed shared clinical features, including hyperuricemia and CKD progression, with phenotypic variability influenced by age and additional genetic modifiers. The findings highlight the prognostic value of genotype-phenotype correlations and the need for tailored clinical management in ADTKD patients.
La enfermedad renal crónica (ERC) refleja una enfermedad grave, asociada a una gran morbimortalidad y creciente gasto presupuestario en salud por paciente. No obstante, a menudo la etiología de esta no está firmemente establecida y, en muchos pacientes, sigue siendo desconocida1. La etiología de la ERC puede ser tanto por factores ambientales como genéticos o la combinación de ambos2,3. Las enfermedades renales dentro del grupo de factores genéticos pueden seguir un modelo de herencia autosómica dominante o recesiva.
Durante años se ha descrito un grupo de enfermedades renales autosómicas dominantes que afecta principalmente a nivel tubular y quístico, caracterizadas por presentar un deterioro progresivo de la función renal acompañada de fibrosis tubulointersticial, siendo UMOD uno de los genes responsables de estas enfermedades.
Presentación de UMOD, función y estructura proteicaEste trabajo está centrado en las manifestaciones fenotípicas causadas por alteraciones en el gen UMOD. Este gen codifica la proteína uromodulina (también conocida como la glucoproteína de Tamm-Horsfall), y es expresada específicamente a nivel renal4.
En condiciones fisiológicas, la uromodulina es la proteína más abundante en orina, y es secretada por las células epiteliales de la rama ascendente gruesa del asa de Henle de manera bidireccional hacia la orina y la circulación4. La uromodulina puede encontrarse polimerizada formando filamentos, o en su forma no polimérica4. Esta forma no polimérica tiene diferentes funciones renales y sistémicas, por lo que es importante su lugar de acción4,5. Las principales funciones de esta proteína son la regulación del transporte iónico en el riñón, la homeostasis urinaria, así como a nivel sistémico como pone de manifiesto Trachtman H6 en su estudio. Adicionalmente, la uromodulina tiene un efecto protector frente la nefrolitiasis por calcio y las infecciones del tracto urinario, con un papel crucial en la inmunomodulación local y sistémica7–9.
Su estructura proteica diferencia 4 dominios similares a EGF (EGF-like I, EGF-like II, EGF-like III y EGF-like IV), un dominio con 10 cistinas conservadas (D10C), y una zona pelúcida (ZP) bipartida en ZP-N y ZP-C10–14. Estos dominios permiten uniones que hacen que la proteína se pliegue tridimensionalmente y forme su estructura funcional y polimerización7,11,12,14,15.
Impacto molecular de las alteraciones genéticas en UMODLas mutaciones en el gen UMOD pueden causar defectos en la estructura, maduración, secreción y/o función de uromodulina, dependiendo del tipo de variante y del dominio o dominios que estén afectados. Gran parte de las alteraciones patogénicas en este gen afectan a la correcta conformación estructural, resultando una proteína anómala. Esta proteína anómala es retenida y acumulada en el retículo endoplásmico, provocando la muerte celular en el túbulo renal7,16,17. Este fenómeno puede desencadenar en estrés del retículo endoplásmico, activación de respuestas inflamatorias, fibrosis intersticial renal y disfunciones mitocondriales secundarias7,18, esto junto a la expresión reducida compensatoria del cotransportador sodio-potasio-cloruro (Na+-K+-2Cl−) (NKCC2)7,13,19, contribuye al incremento en la reabsorción proximal de urato y en el deterioro progresivo de la función renal por el estrés oxidativo que conlleva la acumulación de esta proteína anómala, culminando en hiperuricemia, episodios de gota, fibrosis y fallo renal7,18 (fig. 1).
7. NKCC2: cotransportador sodio-potasio-cloro (Na+-K+-2Cl−); RE: retículo endoplasmático; uUMOD: UMOD en orina.")
Esquema sobre la patofisiología de alteraciones en UMOD. A la izquierda, se representa la restringida expresión de UMOD a la rama ascendente de Henle y las primeras secciones del túbulo contorneado distal. A la derecha, los procesos alterados debido a la expresión de UMOD mutante. Fuente: adaptado de Mabillard, H. et al. (2023)7. NKCC2: cotransportador sodio-potasio-cloro (Na+-K+-2Cl−); RE: retículo endoplasmático; uUMOD: UMOD en orina.
En la literatura se ha asociado una gran variabilidad fenotípica con las alteraciones en UMOD, incluso entre individuos portadores de la misma alteración. No obstante, hay otros factores modificadores genéticos y ambientales que pueden influir en la presentación y progresión de la enfermedad.
Los pacientes portadores de alteraciones en UMOD, a nivel clínico y analítico, pueden presentar una disminución de la excreción fraccionada de urato, lo que provoca hiperuricemia, y afectación glomerular esporádica con manifestaciones como microhematuria. La proteinuria suele ser leve o ausente. También se han observado la aparición de quistes renales en los pacientes con alteraciones en este gen, aunque en mucha menor medida que otras manifestaciones como hiperuricemia20–24.
Por otro lado, se ha observado que los individuos con mutaciones en UMOD alcanzan la enfermedad renal en etapa terminal entre las edades de 25 y 70 años o más, y aquellos con antecedentes de gota experimentaron su aparición entre las edades de 3 y 51 años según el estudio de Ayasreh N et al.25,26.
Los estudios clínicos han demostrado que las biopsias renales de los pacientes afectados revelan fibrosis intersticial y atrofia tubular, lo que confirma el patrón de daño tubulointersticial asociado a estas mutaciones27.
ADTKD-UMOD. Principal enfermedad renal hereditaria asociada a UMODLos genes asociados al desarrollo de nefropatía tubulointersticial autosómica dominante (ADTKD) son HNF1B, MUC1, REN, SEC61A1 y UMOD. Todos manifiestan variabilidad fenotípica entre ellos (diabetes, cáncer hepático, quistes renales y alteraciones psiquiátricas, entre otras), siendo ADTKD la manifestación común que los relaciona, pero con penetrancia incompleta. Las variantes identificadas en el gen UMOD se relacionan mayoritariamente con ADTKD5,7,28–31. Los pacientes con ADTKD-UMOD muestran un curso progresivo de insuficiencia renal con inicio temprano de hiperuricemia y, aunque una gran mayoría de casos se presentan de forma asintomática, también se ha descrito el desarrollo de gota28,30–32.
Mutaciones recurrentes de UMODEstudios recientes en poblaciones cerradas han identificado varias mutaciones recurrentes en UMOD. Estas mutaciones recurrentes son de particular interés debido a su alta prevalencia en poblaciones específicas y su asociación con fenotipos clínicos severos7. El estudio de mutaciones recurrentes ofrece una ventana única para entender los mecanismos patogénicos subyacentes a la función del gen alterado, caracterizar las manifestaciones fenotípicas, estableciendo una correlación entre la mutación genética y la progresión de la enfermedad, y desarrollar estrategias terapéuticas dirigidas.
Se ha descrito una asociación entre la edad de aparición de la enfermedad renal en etapa final, la mutación UMOD subyacente y el dominio en el que se encuentra la mutación32. En este sentido analizar las mutaciones en el gen UMOD y su relación con las enfermedades renales hereditarias proporciona una base sólida para la identificación temprana, el diagnóstico genético y el manejo clínico de estos pacientes.
En los últimos 25 años, nuestro equipo se ha centrado en estudiar y establecer el mapa genético de las enfermedades hereditarias en Galicia dividiendo en 3 grandes grupos: enfermedad quística, enfermedad glomerular y enfermedad tubulointersticial. Este estudio completo engloba 2.538 familias y 4.094 individuos estudiados genéticamente, con el objetivo de establecer una correlación genotipo-fenotipo. Dentro del grupo de enfermedades tubulointersticiales se caracterizaron genéticamente 371 familias y 81 de ellas con ADTKD, de las cuales 31 son familias con mutaciones en el gen UMOD. Dicho estudio identifica la presencia en la población gallega de 2 variantes genéticas recurrentes responsables de la mayoría de los casos de ADTKD sin relación familiar (gen UMOD: p.C255Y y p.Q316P, transcrito NM_001008389.3). En este artículo se establece una correlación genotipo fenotipo en 37 pacientes de 15 familias no relacionadas dentro en la comunidad gallega, con el fin de establecer patrones fenotípicos asociado a la presencia de dichas variantes.
MetodologíaParticipantes del estudio y caracterización clínicaSe trata de un estudio de cohorte retrospectivo, multicéntrico, observacional, en el que se analizó la cohorte NefroCHUS de 4.094 pacientes con sospecha de patologías renales hereditarias. Esta cohorte está formada por los pacientes provenientes de diferentes centros hospitalarios de toda España, con un enriquecimiento en población gallega.
Se seleccionaron aquellos pacientes que cumplían los criterios de inclusión: 1) prueba genética solicitada por un nefrólogo; 2) sospecha clínica inicial de ADTKD; 3) secuenciación genética al menos de los genes asociados a ADTKD (HNF1B, MUC1, REN, SEC61A1 y UMOD); y 4) paciente portador de alguna de las 2 mutaciones más recurrentes del gen UMOD en la comunidad gallega, p.C255Y o p.Q316P.
Se seleccionaron 37 pacientes en total, provenientes de 15 familias. Diez de estas familias portan la alteración p.C255Y, y las 5 restantes, p.Q316P.
Para la caracterización clínica de los pacientes de este estudio se recopilaron datos clínicos como hipertensión, filtrado glomerular estimado (Fge), estadio de ERC, hiperuricemia (considerada cuando los niveles de ácido úrico en suero son mayores de 7mg/dl), excreción fraccional de ácido úrico, episodio de gota, presencia de quistes renales, albuminuria, hematuria, tipo de terapia renal sustitutiva y trasplante renal.
Análisis molecularEl ADN de los pacientes fue extraído de leucocitos de sangre periférica mediante los kits comerciales Chemagic Blood 1k-3k usando el robot de extracción Chemagen (PerkinElmer, Waltham, Massachusetts, EE.UU.) y QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), siguiendo los protocolos recomendados por los fabricantes. La calidad y concentración del ADN extraído se evaluó mediante Qubit 3 fluorometer, kit Broad Range (Thermo Fisher Scientific, Waltham, Massachusetts, EE.UU.).
Todos los pacientes incluidos en esta selección de pacientes se estudiaron genéticamente, ya sea mediante la técnica de secuenciación masiva de ADN, Next-Generation Sequencing (NGS), o mediante secuenciación dirigida, con la técnica de secuenciación Sanger. Siempre que la tecnología lo ha permitido, los casos índices de cada familia han sido estudiados genéticamente con la tecnología NGS (en el 86,67% de los casos índices, 13/15 probandos).
La técnica de NGS empleada en los pacientes de este estudio es la secuenciación de un panel de genes conocidos asociados a las patologías renales tubulares y quísticas más prevalentes33. Con la secuenciación mediante NGS de estos genes se obtiene la secuencia de los exones y regiones flanqueantes. De esta manera se identifican las variantes puntuales, pequeñas inserciones/deleciones (indels) y la presencia de variantes estructurales (Copy Number Variants [CNVs]). Las lecturas de secuenciación crudas se procesaron de acuerdo con las guías de buena práctica de GATK y se alinearon con el genoma humano de referencia (GRCh37) utilizando la versión 0.7.17-r1188. Las lecturas de baja calidad se eliminaron del conjunto de datos primario con la versión 0,20 de fastp. La llamada de variantes se realizó con GATK versión 4.1.9., Pindel versión 0.2.5b9 y ExomeDepth versión 1.1.15, de acuerdo con las recomendaciones de buenas prácticas. La anotación de variantes se llevó a cabo mediante un proceso interno, combinando anotaciones genéticas funcionales de SnpEff y ANNOVAR para recuperar frecuencias poblacionales (1000 Genomes Project, gnomAD y una base de datos interna, entre otras), puntuaciones de predicción funcional (SIFT, CADD, etc.) e información clínica de bases de datos como ClinVar y OMIM.
La secuenciación Sanger es una técnica de secuenciación dirigida en la que se secuencian regiones de interés más cortas de ADN con una alta precisión. Esta técnica es la empleada en la mayoría de los familiares de los casos índices para estudiar la segregación de las alteraciones candidatas. Al identificar la variante candidata de causar patología en el caso índice, se puede dirigir la secuenciación a esa región e identificar de forma más rápida si los familiares son portadores de esta.
Caracterización de las mutaciones identificadasLas variantes fueron clasificadas por 2 genetistas especializados en enfermedades renales hereditarias independientemente siguiendo las guías y recomendaciones del Colegio Americano de Genética Médica (ACMG)34.
Se llevó a cabo una búsqueda bibliográfica para las 2 variantes recurrentes de este estudio usando diferentes herramientas de búsqueda como PubMed, ClinVar, Human Gene Mutation Database (HGMD) y MetaDome con el propósito de determinar si han sido previamente reportadas, su implicación molecular y sus asociaciones fenotípicas.
Análisis estadísticoLa significancia estadística de los resultados se evaluó en variables nominales mediante el test de Chi-cuadrado o test exacto de Fisher para variables con una frecuencia mayor o menor a 5, respectivamente, y para variables cuantitativas, el test Mann-Whitney-Wilcoxon a través de RStudio. Debido al pequeño tamaño muestral, los p-valores se ajustaron usando el método Benjamini-Hochberg para controlar la tasa de falsos descubrimientos (False Discovery Rate [FDR]). Este p-valor ajustado es el q-valor, se considera que alcanza la significación estadística cuando q-valor es mayor de 0,05.
En las curvas de supervivencia de Kaplan-Meier se aplicó el test estadístico de log-rank para estudiar las diferencias entre los tipos de variantes y establecer el p-valor.
ResultadosSe seleccionaron y analizaron 37 pacientes provenientes de 15 familias con Enfermedad Tubulointersticial Renal Autosómica Dominante (ADTKD) portadoras de mutaciones en el gen UMOD, y provenientes de diferentes hospitales de Galicia. Estos pacientes se dividieron en 2 subcohortes. De ellas, 10 probandos y 18 familiares presentan la mutación p.C255Y (todos los individuos portan la alteración en heterocigosis, menos un familiar (F4.S3) que porta la variante en homocigosis), mientras que 5 probandos y 4 familiares portan la mutación p.Q316P (en heterocigosis). Las distribuciones de las edades en ambos grupos son similares (fig. 2A), esto hace que las manifestaciones clínicas de ambas subcohortes sean comparables.
 Distribución de la edad en la cohorte de variantes recurrentes de UMOD dividido por el tipo de variante. Las medianas de la edad en ambos grupos son muy similares, y no se aprecian diferencias estadísticamente significativas entre los 2 grupos (ns), con un p-valor de 0,9222. B) Representación del diagnóstico de hiperuricemia en función de la edad para cada variante mediante una curva de supervivencia Kaplan-Meier, con el test de log-rank se calcula un p-valor de 0,1040, por lo que las diferencias no son estadísticamente significativas. C) Representación de los pacientes en ERCA en función de la edad para cada variante mediante una curva de supervivencia Kaplan-Meier, con el test de log-rank se calcula un p-valor de 0,4156, por lo que las diferencias no son estadísticamente significativas. D) Pacientes en cada estadio de ERC divididas por el tipo de alteración.")
A) Distribución de la edad en la cohorte de variantes recurrentes de UMOD dividido por el tipo de variante. Las medianas de la edad en ambos grupos son muy similares, y no se aprecian diferencias estadísticamente significativas entre los 2 grupos (ns), con un p-valor de 0,9222. B) Representación del diagnóstico de hiperuricemia en función de la edad para cada variante mediante una curva de supervivencia Kaplan-Meier, con el test de log-rank se calcula un p-valor de 0,1040, por lo que las diferencias no son estadísticamente significativas. C) Representación de los pacientes en ERCA en función de la edad para cada variante mediante una curva de supervivencia Kaplan-Meier, con el test de log-rank se calcula un p-valor de 0,4156, por lo que las diferencias no son estadísticamente significativas. D) Pacientes en cada estadio de ERC divididas por el tipo de alteración.
La aparición de hiperuricemia es una de las características más representativas para el diagnóstico clínico de esta enfermedad, es por esto por lo que se estudió la relación de esta aparición en función de la edad para cada una de las variantes mediante una curva de supervivencia Kaplan-Meier (fig. 2B). Al estudiar estadísticamente las diferencias entre las 2 variables por el test de log-rank, se obtuvo un p-valor de 0,1040.
La evolución a ERCA es una de las características de los pacientes con alteraciones en UMOD, debido al deterioro progresivo de la función renal. Es por esto que, en la figura 2C se puede apreciar que la ERCA no se da en ningún caso menor de 38 años, y a partir de esta edad hay un continuo descenso de la curva de supervivencia para ambas alteraciones.
La figura 2D representa la distribución de los diferentes estadios de ERC en cada subcohorte. A pesar de que no se encuentran diferencias estadísticamente significativas para el estadio de ERC (tabla 1), en la figura 2D se observa que, mientras que los pacientes portadores de la variante p.C255Y tienen una mayor frecuencia en el estadio 3b, los portadores de p.Q316P tienen menor representación en este estadio. Por lo que, sí que podría haber una tendencia con respecto a la progresión de la enfermedad.
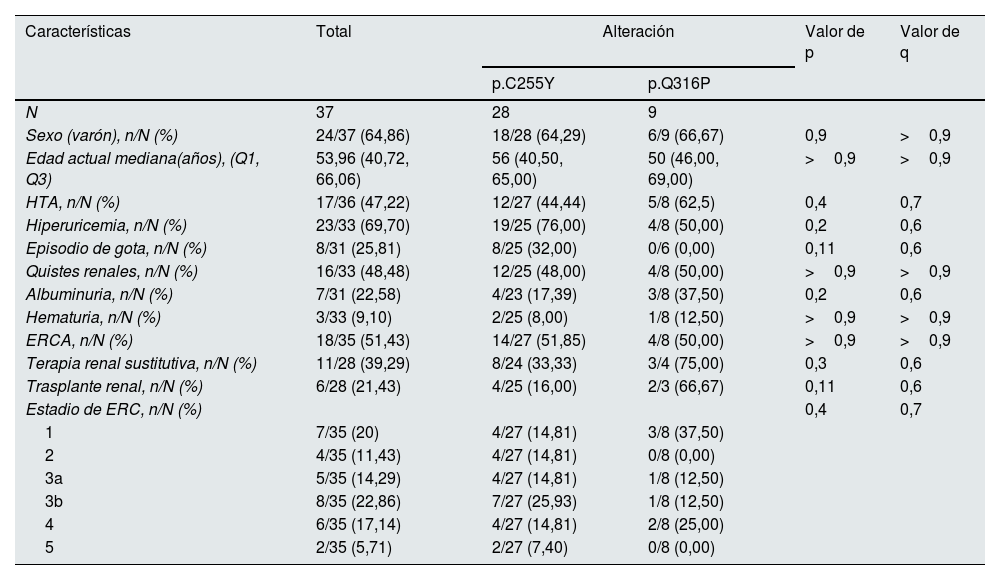
Descripción estadística de las manifestaciones clínicas de los pacientes con alteraciones recurrentes en UMOD en función de su alteración
| Características | Total | Alteración | Valor de p | Valor de q | |
|---|---|---|---|---|---|
| p.C255Y | p.Q316P | ||||
| N | 37 | 28 | 9 | ||
| Sexo (varón), n/N (%) | 24/37 (64,86) | 18/28 (64,29) | 6/9 (66,67) | 0,9 | >0,9 |
| Edad actual mediana(años), (Q1, Q3) | 53,96 (40,72, 66,06) | 56 (40,50, 65,00) | 50 (46,00, 69,00) | >0,9 | >0,9 |
| HTA, n/N (%) | 17/36 (47,22) | 12/27 (44,44) | 5/8 (62,5) | 0,4 | 0,7 |
| Hiperuricemia, n/N (%) | 23/33 (69,70) | 19/25 (76,00) | 4/8 (50,00) | 0,2 | 0,6 |
| Episodio de gota, n/N (%) | 8/31 (25,81) | 8/25 (32,00) | 0/6 (0,00) | 0,11 | 0,6 |
| Quistes renales, n/N (%) | 16/33 (48,48) | 12/25 (48,00) | 4/8 (50,00) | >0,9 | >0,9 |
| Albuminuria, n/N (%) | 7/31 (22,58) | 4/23 (17,39) | 3/8 (37,50) | 0,2 | 0,6 |
| Hematuria, n/N (%) | 3/33 (9,10) | 2/25 (8,00) | 1/8 (12,50) | >0,9 | >0,9 |
| ERCA, n/N (%) | 18/35 (51,43) | 14/27 (51,85) | 4/8 (50,00) | >0,9 | >0,9 |
| Terapia renal sustitutiva, n/N (%) | 11/28 (39,29) | 8/24 (33,33) | 3/4 (75,00) | 0,3 | 0,6 |
| Trasplante renal, n/N (%) | 6/28 (21,43) | 4/25 (16,00) | 2/3 (66,67) | 0,11 | 0,6 |
| Estadio de ERC, n/N (%) | 0,4 | 0,7 | |||
| 1 | 7/35 (20) | 4/27 (14,81) | 3/8 (37,50) | ||
| 2 | 4/35 (11,43) | 4/27 (14,81) | 0/8 (0,00) | ||
| 3a | 5/35 (14,29) | 4/27 (14,81) | 1/8 (12,50) | ||
| 3b | 8/35 (22,86) | 7/27 (25,93) | 1/8 (12,50) | ||
| 4 | 6/35 (17,14) | 4/27 (14,81) | 2/8 (25,00) | ||
| 5 | 2/35 (5,71) | 2/27 (7,40) | 0/8 (0,00) | ||
Los valores de p comparan las 2 alteraciones recurrentes usando en datos cuantitativos el test de Mann-Whitney-Wilcoxon y para los datos categóricos el de Chi-cuadrado o el test exacto de Fisher para variables con una frecuencia de más o menos 5, respectivamente; los valores de q son los valores de p corregidos con el test de Benjamini-Hochberg (corrección de la tasa de falsos descubrimientos para pruebas múltiples). ERCA: enfermedad renal crónica avanzada; HTA: hipertensión arterial; n: representa el número de sujetos con la característica; N: representa el número de sujetos totales con información sobre esa característica.
La tabla 1 representa la estadística descriptiva de las características observadas en los pacientes de esta cohorte junto con sus proporciones, mientras la tabla 2 detalla las características observadas en cada paciente.
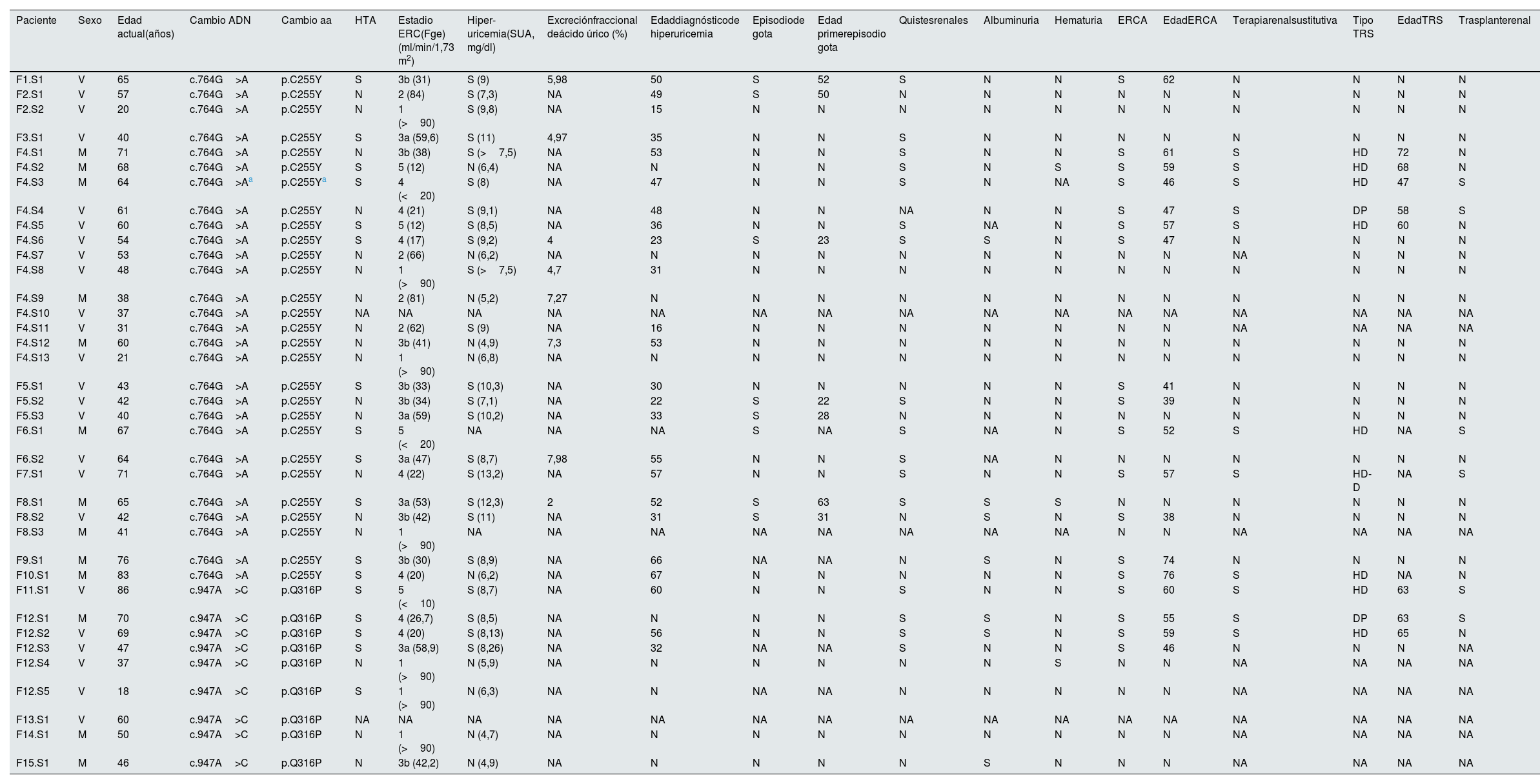
Características fenotípicas de la cohorte de pacientes con alteraciones recurrentes en UMOD
| Paciente | Sexo | Edad actual(años) | Cambio ADN | Cambio aa | HTA | Estadio ERC(Fge) (ml/min/1,73 m2) | Hiper-uricemia(SUA, mg/dl) | Excreciónfraccional deácido úrico (%) | Edaddiagnósticode hiperuricemia | Episodiode gota | Edad primerepisodio gota | Quistesrenales | Albuminuria | Hematuria | ERCA | EdadERCA | Terapiarenalsustitutiva | Tipo TRS | EdadTRS | Trasplanterenal |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1.S1 | V | 65 | c.764G>A | p.C255Y | S | 3b (31) | S (9) | 5,98 | 50 | S | 52 | S | N | N | S | 62 | N | N | N | N |
| F2.S1 | V | 57 | c.764G>A | p.C255Y | N | 2 (84) | S (7,3) | NA | 49 | S | 50 | N | N | N | N | N | N | N | N | N |
| F2.S2 | V | 20 | c.764G>A | p.C255Y | N | 1 (>90) | S (9,8) | NA | 15 | N | N | N | N | N | N | N | N | N | N | N |
| F3.S1 | V | 40 | c.764G>A | p.C255Y | S | 3a (59,6) | S (11) | 4,97 | 35 | N | N | S | N | N | N | N | N | N | N | N |
| F4.S1 | M | 71 | c.764G>A | p.C255Y | N | 3b (38) | S (>7,5) | NA | 53 | N | N | S | N | N | S | 61 | S | HD | 72 | N |
| F4.S2 | M | 68 | c.764G>A | p.C255Y | S | 5 (12) | N (6,4) | NA | N | N | N | S | N | S | S | 59 | S | HD | 68 | N |
| F4.S3 | M | 64 | c.764G>Aa | p.C255Ya | S | 4 (<20) | S (8) | NA | 47 | N | N | S | N | NA | S | 46 | S | HD | 47 | S |
| F4.S4 | V | 61 | c.764G>A | p.C255Y | N | 4 (21) | S (9,1) | NA | 48 | N | N | NA | N | N | S | 47 | S | DP | 58 | S |
| F4.S5 | V | 60 | c.764G>A | p.C255Y | S | 5 (12) | S (8,5) | NA | 36 | N | N | S | NA | N | S | 57 | S | HD | 60 | N |
| F4.S6 | V | 54 | c.764G>A | p.C255Y | S | 4 (17) | S (9,2) | 4 | 23 | S | 23 | S | S | N | S | 47 | N | N | N | N |
| F4.S7 | V | 53 | c.764G>A | p.C255Y | N | 2 (66) | N (6,2) | NA | N | N | N | N | N | N | N | N | NA | N | N | N |
| F4.S8 | V | 48 | c.764G>A | p.C255Y | N | 1 (>90) | S (>7,5) | 4,7 | 31 | N | N | N | N | N | N | N | N | N | N | N |
| F4.S9 | M | 38 | c.764G>A | p.C255Y | N | 2 (81) | N (5,2) | 7,27 | N | N | N | N | N | N | N | N | N | N | N | N |
| F4.S10 | V | 37 | c.764G>A | p.C255Y | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| F4.S11 | V | 31 | c.764G>A | p.C255Y | N | 2 (62) | S (9) | NA | 16 | N | N | N | N | N | N | N | NA | NA | NA | NA |
| F4.S12 | M | 60 | c.764G>A | p.C255Y | N | 3b (41) | N (4,9) | 7,3 | 53 | N | N | N | N | N | N | N | N | N | N | N |
| F4.S13 | V | 21 | c.764G>A | p.C255Y | N | 1 (>90) | N (6,8) | NA | N | N | N | N | N | N | N | N | N | N | N | N |
| F5.S1 | V | 43 | c.764G>A | p.C255Y | S | 3b (33) | S (10,3) | NA | 30 | N | N | N | N | N | S | 41 | N | N | N | N |
| F5.S2 | V | 42 | c.764G>A | p.C255Y | N | 3b (34) | S (7,1) | NA | 22 | S | 22 | S | N | N | S | 39 | N | N | N | N |
| F5.S3 | V | 40 | c.764G>A | p.C255Y | N | 3a (59) | S (10,2) | NA | 33 | S | 28 | N | N | N | N | N | N | N | N | N |
| F6.S1 | M | 67 | c.764G>A | p.C255Y | S | 5 (<20) | NA | NA | NA | S | NA | S | NA | N | S | 52 | S | HD | NA | S |
| F6.S2 | V | 64 | c.764G>A | p.C255Y | S | 3a (47) | S (8,7) | 7,98 | 55 | N | N | S | NA | N | N | N | N | N | N | N |
| F7.S1 | V | 71 | c.764G>A | p.C255Y | N | 4 (22) | S (13,2) | NA | 57 | N | N | S | N | N | S | 57 | S | HD-D | NA | S |
| F8.S1 | M | 65 | c.764G>A | p.C255Y | S | 3a (53) | S (12,3) | 2 | 52 | S | 63 | S | S | S | N | N | N | N | N | N |
| F8.S2 | V | 42 | c.764G>A | p.C255Y | N | 3b (42) | S (11) | NA | 31 | S | 31 | N | S | N | S | 38 | N | N | N | N |
| F8.S3 | M | 41 | c.764G>A | p.C255Y | N | 1 (>90) | NA | NA | NA | NA | NA | NA | NA | NA | N | N | NA | NA | NA | NA |
| F9.S1 | M | 76 | c.764G>A | p.C255Y | S | 3b (30) | S (8,9) | NA | 66 | NA | NA | N | S | N | S | 74 | N | N | N | N |
| F10.S1 | M | 83 | c.764G>A | p.C255Y | S | 4 (20) | N (6,2) | NA | 67 | N | N | N | N | N | S | 76 | S | HD | NA | N |
| F11.S1 | V | 86 | c.947A>C | p.Q316P | S | 5 (<10) | S (8,7) | NA | 60 | N | N | S | N | N | S | 60 | S | HD | 63 | S |
| F12.S1 | M | 70 | c.947A>C | p.Q316P | S | 4 (26,7) | S (8,5) | NA | N | N | N | S | S | N | S | 55 | S | DP | 63 | S |
| F12.S2 | V | 69 | c.947A>C | p.Q316P | S | 4 (20) | S (8,13) | NA | 56 | N | N | S | S | N | S | 59 | S | HD | 65 | N |
| F12.S3 | V | 47 | c.947A>C | p.Q316P | S | 3a (58,9) | S (8,26) | NA | 32 | NA | NA | S | N | N | S | 46 | N | N | N | NA |
| F12.S4 | V | 37 | c.947A>C | p.Q316P | N | 1 (>90) | N (5,9) | NA | N | N | N | N | N | S | N | N | NA | NA | NA | NA |
| F12.S5 | V | 18 | c.947A>C | p.Q316P | S | 1 (>90) | N (6,3) | NA | N | NA | NA | N | N | N | N | N | NA | NA | NA | NA |
| F13.S1 | V | 60 | c.947A>C | p.Q316P | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| F14.S1 | M | 50 | c.947A>C | p.Q316P | N | 1 (>90) | N (4,7) | NA | N | N | N | N | N | N | N | N | NA | NA | NA | NA |
| F15.S1 | M | 46 | c.947A>C | p.Q316P | N | 3b (42,2) | N (4,9) | NA | N | N | N | N | S | N | N | N | NA | NA | NA | NA |
DP: diálisis peritoneal; ERC: enfermedad renal crónica; ERCA: enfermedad renal crónica avanzada; Fge: filtrado glomerular estimado (calculado con CKD-EPI); HD: hemodiálisis; HD-D: hemodiálisis domiciliaria; M: mujer; N: no; NA: no hay datos disponibles; S: sí; SUA: ácido úrico en suero; TRS: terapia renal sustitutiva; V: varón.
A nivel molecular la alteración p.C255Y está clasificada como probablemente patogénica. Esta afecta al dominio D10C10, este dominio se encuentra altamente conservado, por lo que las alteraciones genéticas que lo afectan tienen alta probabilidad de tener un impacto en su función13,35,36. Al estudiar la tolerancia al cambio del aminoácido de referencia en la posición 255 con MetaDome37, vemos que este es intolerante al cambio, con una puntuación de tolerancia de 0,22 (las puntuaciones menores de 0,7 son consideradas intolerantes al cambio de aminoácido), por lo tanto, esta alteración recurrente que cambia el residuo de cistina por el de tirosina, tiene un impacto negativo a nivel proteico37.
Esta es la variante más recurrente en nuestra cohorte gallega de pacientes con sospecha de enfermedad renal hereditaria, con un total de 28 pacientes portadores. De estos, el 64,29% son varones (18/28).
Doce pacientes presentan hipertensión arterial (HTA) (44,44%), ya sea en el momento del diagnóstico o en el transcurso de la enfermedad. Además, el 77,77% de los pacientes mostraba algún grado de ERC (considerado a partir de estadio 2, incluido) al momento del estudio, con una media de 57 años. Ocho pacientes (33,33%) han progresado a ERCT, iniciando diálisis a una edad promedio de 61 años.
Destaca el caso de la paciente F4.S3, portadora de la alteración p.C255Y en homocigosis. F4.S3 comenzó terapia renal sustitutiva (TRS) a los 46 años, una década o más de adelanto que el resto de los pacientes con TRS de la cohorte (tabla 2). Esto sugiere que las alteraciones en homocigosis se asocian con un progreso más acelerado y severo de la enfermedad renal.
Un 76% de los casos cumplieron los criterios de hiperuricemia (19 de 25 pacientes). El diagnóstico de hiperuricemia e inicio de tratamiento con alopurinol se realizó a una edad media de 41,38 años (rango: 16-67 años). El sedimento urinario fue generalmente anodino, con solo 4 casos de albuminuria (17,39%) y 2 casos de microhematuria (8%). Además, se observó desarrollo de quistes en el 48% de los pacientes.
Dentro del 32% de los pacientes con gota, la edad media en la que apareció el primer episodio de gota es 38,43 años (rango: 22-63 años). En la figura 3 se puede apreciar la curva de supervivencia de las edades asociadas al primer episodio de gota. A pesar de comenzar en edades adultas tempranas, hay varios pacientes de avanzada edad que no han desarrollado gota, lo que podría interpretarse como que este evento no está altamente asociado a la edad.
Variabilidad intrafamiliar de p.C255Y. Familia F4
De estos pacientes portadores, 13 pertenecen a la familia F4, esta presenta antecedentes de hiperuricemia y Enfermedad Renal Crónica Terminal (ERCT), condiciones que, probablemente, están asociadas a esta mutación. 2 personas de esta familia han tenido un trasplante renal. A nivel intrafamiliar observamos características con una gran prevalencia familiar, como un filtrado glomerular estimado menor de 90ml/min/1,73 m2 indicativo de enfermedad renal.
Adicionalmente se ha encontrado variabilidad en cuanto al desarrollo de hiperuricemia, observándose esta característica en los pacientes de distintas edades.
También se observa cierta variabilidad asociada a la edad en cuanto a otras características, en las que aproximadamente la mitad de los familiares con mayor edad presentan la característica y la otra mitad de familiares más jóvenes, no. Siendo estas características: la llegada a enfermedad renal crónica avanzada (ERCA) (los individuos menores de 54 años en esta familia no han progresado a ERCA), TRS (en esta característica la edad de corte son los 60 años), presencia de quistes renales (con una edad de corte de 54 años).
Cabe destacar también que 6 miembros de esta familia son además portadores de una alteración patogénica en heterocigosis del gen PKHD1, c.383delC, p.Thr128fs (F4.S1, F4.S2, F4.S6, F4.S7, F4.S9 y F4.S13). Alteraciones en este gen están asociadas con principalmente con el desarrollo de quistes renales con una herencia principalmente autosómica recesiva, aunque se ha descrito fenotipo quístico también en los pacientes con alteraciones en heterocigosis38. En la familia F4 hay un total de 5 individuos con quistes renales, de los cuales 3 de ellos son portadores de la alteración en PKHD1. Es por esto que no podemos confirmar ni descartar que esta alteración en PKHD1 module la aparición de quistes renales en los pacientes portadores de alteraciones en UMOD y PKHD1.
Alteración UMOD c.947A>C (p.Q316P)Por otro lado, la alteración p.Q316P también es clasificada molecularmente como probablemente patogénica. Afecta al domino EGF-like IV10–12, este dominio está altamente conservado e interviene en las uniones bisulfato que contribuyen a la estructura tridimensional de la uromodulina10. Esta posición también es intolerante al cambio con una puntuación de tolerancia de 0,3637.
Esta variante la encontramos en 9 pacientes, de los cuales el 66,67% son varones (6/9). En esta cohorte se observó un mayor porcentaje de pacientes con HTA, presente en 5 pacientes (62,5%). Al momento del diagnóstico, el 50% de los pacientes presentaba cierto grado de ERC (considerado a partir del estadio 2, incluido), a una edad media de 54 años. Hasta la fecha, 3 pacientes (75%) han alcanzado la ERCT, con inicio de TRS a una edad media de 63,67 años.
El 50% de la cohorte cumplió criterios de hiperuricemia. El diagnóstico de hiperuricemia y el inicio de tratamiento con alopurinol se dieron a una edad media de 49,33 años (rango: 32-60 años). Hasta el momento, no se han registrado episodios de crisis gotosa en estos pacientes, aunque sí existen antecedentes familiares de gota.
En cuanto al sedimento urinario de esta segunda cohorte, fue también anodino, con microalbuminuria presente en 3 de los 8 pacientes (uno de ellos con CAC>300mg/g) y solo un caso de microhematuria. De manera similar a la primera subcohorte, se observó desarrollo quístico en el 50% de los pacientes.
Variabilidad intrafamiliar de p.Q316P. Familia F12La familia F12 tiene el mayor número de integrantes portadores de p.Q316P, consta de 5 pacientes, y 4 de estos presentan HTA. En esta familia se aprecia también el efecto de la edad asociado al deterioro renal, ya que los 2 integrantes más jóvenes de la familia solo presentan fenotipo patológico en cuanto a HTA, sin ninguna característica adicional, mientras que los pacientes de más edad presentan un filtrado glomerular de 35,2ml/min/1,73m2; quistes renales; hiperuricemia; y han alcanzado ERCA.
DiscusiónEste es un estudio retrospectivo sobre las variantes recurrentes en UMOD de una cohorte de población gallega cerrada desde el punto de vista genético. El estudio de la población gallega es de un alto valor en cuanto a la caracterización de variantes recurrentes y de correlaciones genotipo-fenotipo, ya que la región de Galicia se encuentra geográficamente localizada en el oeste de España, y barreras geográficas han proporcionado un alto nivel de conservación genética, etnicidad y consanguinidad39,40.
Las alteraciones estudiadas se encuentran localizadas en posiciones muy poco tolerantes al cambio37, altamente conservadas10–12 y con una función estructural10,13,35,36, es por esto por lo que, aunque se trata de una alteración que cambia únicamente un aminoácido por otro, las variantes tienen un efecto patogénico, y resultan en manifestaciones renales en los pacientes, ya que la proteína no puede estructurarse tridimensionalmente de manera correcta, y su función se ve alterada.
Con los datos clínicos obtenidos de esta cohorte, es notable que estas alteraciones en el gen UMOD tienen ciertos fenotipos comunes, confirmando el importante papel del gen en procesos como el transporte y absorción de ácido úrico, formación de quistes y función renales, que puede derivar con los años a un deterioro renal temprano.
De toda la cohorte, tan solo F4.S3 porta la alteración p.C255Y en homocigosis, tanto en este estudio como en otros anteriores se ha observado que las alteraciones bialélicas en este gen producen un desarrollo más agresivo y temprano de las manifestaciones clínicas41–45.
A pesar de la diferencia de tamaño de las 2 subcohortes, se pueden apreciar ciertas similitudes y diferencias entre los pacientes portadores de estas. Dentro de las semejanzas, y en consonancia con la literatura sobre el fenotipo asociado a las alteraciones en UMOD, más de la mitad de los pacientes presentan hiperuricemia (76 y 50%, respectivamente). Se observa presencia de quistes renales y HTA en aproximadamente la mitad de los portadores para ambas alteraciones. Aproximadamente la mitad de los pacientes llegaron a ERCA (con mayor representación en la población de edad más avanzada), y han necesitado un tratamiento renal sustitutivo.
El estudio de Moskowitz JL et al.32 compara el fenotipo de varias alteraciones en UMOD, incluidas las alteraciones, p.C255Y y p.Q316P. Estos pacientes presentan principalmente hiperuricemia, gota (en menor proporción) y llegan a enfermedad renal terminal. En este estudio se observó que la alteración con la que los pacientes llegaban más tarde a enfermedad renal terminal era p.Q316P32. En nuestra cohorte no se aprecian diferencias estadísticas entre las alteraciones. Aun así (fig. 2C), los portadores de p.Q316P tardan más en llegar a ERCA, siendo el primer caso a los 46 años, mientras que el primer caso de los portadores de p.C255Y, fue a los 38 años. Cabe destacar que esta característica está relacionada con el deterioro renal debido a múltiples factores y asociado directamente con la edad.
En la figura 2B se puede apreciar que la presencia de hiperuricemia es una característica que puede darse en distintas etapas de la vida, en algunos pacientes tuvo una aparición en etapas tempranas, mientras que, en otros, se diagnosticó a una edad más avanzada. Y a pesar de que hay diferencias en el tamaño de las subcohortes, se observa cierta tendencia con un p-valor cercano a la significancia (0,1040) a que la variante p.C255Y está asociada con una anterior aparición de hiperuricemia en los pacientes.
En la tabla 1 no hay diferencias estadísticamente significativas entre estas variantes recurrentes, pero aun así se aprecian cierta tendencia de los pacientes con p.C255Y a tener estadios mayores de ERC, mientras que el 37,5% de los portadores de p.Q316P, continúan con un flujo glomerular estimado normal (estadio ERC 1) (fig. 2D).
Es conocido que el dominio D10C es crucial para mantener la correcta estructura tridimensional de la proteína, de hecho, el grupo de Ma L et al.13 estudiaron las diferencias in vivo entre una alteración dentro del dominio (p.C217G) y fuera de este (p.C126R), concluyendo que cuando el dominio D10C se ve alterado, tiene mayores efectos deletéreos a nivel apoptótico, de retención en el retículo endoplasmático, se reducía la translocación a la superficie apical y su liberación extracelular13,15. Esto coincide con lo observado en nuestra cohorte, en la que los pacientes con la alteración en este dominio están en estadios de ERC mayores (fig. 2D).
Los datos disponibles muestran un mayor porcentaje de pacientes portadores de p.Q316P sometidos a TRS y a trasplante renal (tabla 1). Estas altas proporciones puede deberse a que hay pocos pacientes con información al respecto de estas características, por lo que los porcentajes pueden sobreestimarse.
Es destacable que, siendo la fracción de excreción de ácido úrico relevante, esta no suele solicitarse de forma rutinaria en los pacientes teniendo información en 8 de los 37 pacientes.
También cabe destacar a nivel clínico que el tipo de TRS más empleado en los pacientes es la hemodiálisis, usada en 9 pacientes en total de esta cohorte (7 de ellos portan la alteración p.C255Y, y 2, p.Q316P). Por otro lado, la diálisis peritoneal es la empleada en 2 pacientes, uno de cada tipo de alteración.
El desarrollo de hiperuricemia ha sido la característica con mayor variabilidad intrafamiliar en ambas alteraciones. También se observan diferencias entre aquellos familiares más jóvenes y mayores, habiendo desarrollado estos últimos ciertas características asociadas a la edad como quistes o deterioro de la función renal, siendo probable que los individuos más jóvenes desarrollen esta sintomatología con el tiempo (especialmente en la familia F12). Estos datos aportan información pronóstica asociada a estas variantes recurrentes. Pero esta correlación con la edad no se observa en todos los casos, por ejemplo, F8.S2 presenta una sintomatología más severa y temprana que F8.S1, a pesar de ser más joven.
En ambas subcohortes, el interrogatorio guiado reveló síntomas como polidipsia y poliuria, aunque parece estar subestimado el aumento en el volumen urinario y la mayor ingesta hídrica, debido a la baja autopercepción de estos síntomas, que han sido constantes en la vida de estos pacientes.
La principal debilidad de este estudio es que, a pesar de que las 2 subcohortes están formadas por pacientes con edades comparables, la diferencia de tamaño entre estas hace que no se pueda hacer una comparación correcta en ciertas características con menos información.
Entre los puntos fuertes del estudio destaca la observación clínica de 2 variantes recurrentes en una cohorte cerrada. Esto aporta y enriquece el conocimiento sobre sus características al reunir un gran número de pacientes portadores de la misma alteración genética. Otro punto fuerte es el detallado fenotipaje de los pacientes, gracias al cual se obtiene valiosa información clínica que permite ahondar en este conocimiento sobre las variantes recurrentes.
Con todo esto se puede relacionar la manifestación de características como el desarrollo de hiperuricemia (llegando en algunos casos a tener episodios de gota), quistes renales y deterioro de la función renal con alteraciones en el gen UMOD. Incidiendo en las alteraciones recurrentes estudiadas, se ha observado que los pacientes portadores de p.C255Y tienden a presentar hiperuricemia a edades más temprana que los portadores de p.Q316P y solo presentaron episodios de gota los portadores de p.C255Y. A pesar de que no se aprecia ninguna tendencia en cuanto a una edad más temprana o tardía de llegada a ERCA, se observa una mayor incidencia de los portadores de p.C255Y en estadios 3a, 3b y 4. Estas diferencias podrían ser debidas a que la estructura proteica se ve alterada en diferentes grados, ya que las alteraciones se encuentran en distintos dominios.
La correlación genotipo-fenotipo de este estudio es de gran importancia pronóstica al proporcionar información sobre cómo podría evolucionar la patología renal en pacientes portadores de estas variantes recurrentes. Como perspectiva futura, se podría hacer una comparación de otras variantes en UMOD, y así conocer las particularidades que pueda presentar cada una. O incluso, ampliar el estudio a variantes en otros genes asociados a ADTKD, comparando el fenotipo de los pacientes, y estableciendo nuevas correlaciones genotipo-fenotipo para esta enfermedad.
FinanciaciónEste estudio ha sido financiado por el Instituto de Salud Carlos III (ISCIII) (fondo REDinREN RD016/0009, a M.Á.G.G.; RICORS2040 RD21/0005/0020 y RD24/0004/0010 financiado por la Unión Europea-NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR) a M.Á.G.G.; y proyectos PI15/01467, PI18/0037 y PI22/00227 cofinanciados por la Unión Europea. a M.Á.G.G.); la Fundación Senefro (SEN2021_2 y SEN2024 a M.Á.G.G.); la Xunta de Galicia/GAIN (Grupos con potencial de crecemento-GPC fondo IN607B 2016/020 e IN607B 2023/07 a M. Á.G.G.); y apoyo salarial de la Xunta de Galicia (Becas predoctorales, a E.S.C.) y del ISCIII a través del PMP21-000109 (a M.G.M. y E.S.C.).
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.
Los autores expresan su gratitud al PKDcore por proporcionar materiales de investigación que contribuyen a ampliar el conocimiento sobre las enfermedades renales y a la investigación traslacional, también les gustaría agradecer a la asociación Art for Dent por sus donaciones al grupo de investigación.
Dr. Alfonso Otero González (CHUOU): 7 pacientes.
Dr. Jesus Angel Calviño Varela (HULA): 4 pacientes.
Dr. Alfredo Reparaz Andrade (CHUVI): 3 pacientes.
Dr. Borja Temes Álvarez (CHUOU): 2 pacientes.
Dra. Beatriz Millán Díaz (HULA): 2 pacientes.
Dra. Ana Isabel Díaz Mareque (CHUOU/CHUS): 2 pacientes.
Dra. Carmen Vázquez Gómez (CHUS): 2 pacientes.
Dra. Mª Jesús Camba Caride (CHUOU): 2 pacientes.
Dr. Cándido Díaz Rodríguez (CHUS): un paciente.
Dr. Juan José Bravo Lopez (CHUOU): un paciente.
Dra. Adela Urisarri Ruiz de Cortaza (CHUS): un paciente.
Dra. Alba Rivas Oural (CHUOU): un paciente.
Dra. Ana Rodríguez-Carmona de la Torre (CHUAC): un paciente.
Dra. Araceli García Pose (HULA): un paciente.
Dra. Carmen Cobelo Casas (HULA): un paciente.
Dra. Diana Feijoo Piñeiro (CHOP): un paciente.
Dra. Elena Iglesias Lamas (CHUOU): un paciente.
Dra. Luisa Palomares Solla (CHUVI): un paciente.
Dra. Olaia Conde Rivera (CHOP): un paciente.
Dr. Miguel Pérez Fontán (CHUAC): un paciente.
Dra. María Jesús Castro Vilanova (CHOP): un paciente.
recomendados


AL DÍA

