




Secondary hyperoxaluria is a metabolic disorder characterized by an increase in urinary oxalate excretion. The etiology may arise from an increase in the intake of oxalate or its precursors, decreased elimination at the digestive level, or heightened renal excretion. Recently, the role of the SLC26A6 transporter in the etiopathogenesis of this disease has been identified. This transporter is active at both the intestinal and renal levels, and its mechanism of action is disrupted during systemic inflammation and metabolic syndrome, which could explain the rising incidence of secondary hyperoxaluria in recent decades. Treatment includes hygienic dietary measures, and medications aimed at reducing intestinal absorption by increasing fecal excretion. Different immunomodulatory drugs, microbiome modifiers and SGLT2 inhibitors could constitute new therapeutic targets. Currently, specific treatments for secondary hyperoxaluria are lacking, making early diagnosis and preventive measures against kidney failure the main therapeutic strategies.
La hiperoxaluria secundaria es un trastorno metabólico caracterizado por un aumento en la excreción urinaria de oxalato. La etiología puede deberse a un aumento de la ingesta de oxalato o sus precursores, una disminución en la eliminación a nivel digestivo o un aumento en la excreción renal. Recientemente se ha descubierto el papel del transportador SLC26A6 en la etiopatogenia de esta enfermedad, presente tanto a nivel intestinal como renal, su mecanismo de acción se ve alterado en situaciones de inflamación sistémica y síndrome metabólico, lo que podría explicar el aumento creciente de los casos de hiperoxaluria secundaria en las últimas décadas. El tratamiento incluye medidas higiénico dietéticas, así como fármacos orientados a disminuir su absorción a nivel intestinal aumentando la excreción fecal. Diferentes fármacos inmunomoduladores, modificadores del microbioma y los inhibidores del SGLT2 podrían constituir nuevas dianas terapéuticas. En el momento actual no disponemos de tratamientos específicos para la hiperoxaluria secundaria, por lo que el diagnóstico precoz y las medidas orientadas a prevenir el avance de la insuficiencia renal son actualmente las principales herramientas terapéuticas.
We present the case of a 69-year-old man admitted for acute renal failure “Acute Kidney Injury Network (AKIN 3)” in the context of diarrhea.
The patient had a personal history of hypertension and type 2 diabetes mellitus lasting over 20 years, with micro-macrovascular involvement, managed with good metabolic control on insulin treatment; diabetic kidney disease stage 3bA1 (baseline creatinine of 1.5 mg/dL and estimated glomerular filtration rate of 35 mL/min/1.73 m2) and obesity, which was treated with bariatric surgery in 2013. Additionally, in the weeks prior to admission, he was diagnosed with immunoglobulin G (IgG) kappa multiple myeloma and was undergoing treatment with daratumumab, bortezomib, melphalan, and prednisone.
At the time of admission, the patient had received his second cycle of chemotherapy, achieving partial hematologic remission. Following this cycle, he experienced profuse diarrhea, and acute renal injury was noted, with peak creatinine of 6 mg/dL and urea of 200 mg/dL, initially attributed to a functional reduction in renal function. Since melphalan could be responsible for both the diarrhea and direct renal damage, it was decided to discontinue this medication. After resolution of this condition, the patient was discharged with a creatinine level of 1.9 mg/dL.
Six months later, he experienced a new episode of acute kidney injury of unknown etiology (peak creatinine of 5.1 mg/dL), showing no improvement despite adjustments to blood volume and requiring renal replacement therapy. Once the progression or renal involvement due to hematologic pathology was ruled out, a renal biopsy was performed, revealing an increase in the glomerular mesangial matrix, suggesting incipient diabetic nephropathy, along with the presence of transparent crystalline structures located in the lumen of the proximal tubules. These birefringent crystals did not show deposits in the immunofluorescence study, compatible with oxalate crystal involvement (Fig. 1).

Histopathological findings. A) Histological evaluation performed on a kidney cylinder with including six glomeruli, of which two present global sclerosis and two have pericapsular fibrosis. The interstitium presented moderate fibrosis (40%) accompanied by tubular atrophy (20%). B) Glomerulus with diffuse increase in the mesangial matrix and thickening of the capillary wall, without mesangial hypercellularity. Hyaline arteriolosclerosis. C) Irregular and translucent crystals, located in the tubular lumen and within the cytoplasm of the tubular epithelial cells. The tubular epithelium showed signs of acute tubular damage with regenerative changes. Chronic inflammatory infiltrate was identified in the interstitium. The crystals were markedly birefringent with polarized light.
Given the irreversibility of the lesions, the patient is currently on chronic hemodialysis.
IntroductionHyperoxaluria is a metabolic disorder with increasing incidence in which there is an increased excretion of urinary oxalate. Excess oxalate may be due to inherited enzyme defects that lead to hepatic overproduction of oxalate (primary hyperoxaluria) or to increased intestinal absorption of oxalate (secondary hyperoxaluria). Oxalate is eliminated unmetabolized by the kidneys;thus,in hyperoxaluria the kidneys are the first organs affected, leading to nephrocalcinosis, lithiasis, and renal failure. It can also be deposited in all organs and tissues except the liver, a condition known as oxalosis.1–7
The aim of the present review is to analyze, in depth and guided by a clinical case, the current situation of secondary hyperoxaluria as the most common form of hyperoxaluria.
PathophysiologyOxalic acid (C2O4H2) (molecular weight 90 Da) is an anion with no apparent function in the human organism. However which, in the plant world, it performs the function of support by forming part of the exoskeleton.8 Its main source is endogenous, derived from the metabolism of ascorbic acid and glyoxylic acid or glyoxylate, although a small part comes directly from the diet.9
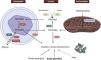
Glyoxylate is a molecule generated during the intermediate metabolism of glycine, hydroxyproline, and glycolate, whose detoxification is primarily conducted by alanine-glyoxylate aminotransferase (AGT) in the peroxisome of the human hepatocyte, converting glyoxylate into glycine. In this reaction, vitamin B6 acts as a cofactor. In healthy individuals, only a portion of the glyoxylate is converted to oxalate by lactate dehydrogenase (LDHA), while the remainder is metabolized by glyoxylate reductase-hydroxypyruvate reductase (GRHPR) to glycolate1,8,10,11 (Fig. 2).

Hepatic metabolism of oxalate. Glycine is metabolized to glyoxylate by the enzyme alanine-glyoxylate aminotransferase (AGT) in the human hepatocyte. In healthy subjects, most glyoxylate is metabolized in the cytosol to glycolate via the enzyme glyoxylate reductase-hydroxypyruvate reductase (GRHPR); only a small amount is metabolized by lactate dehydrogenase (LDHA) to oxalate.
ADH: alcohol dehydrogenase; AGT: alanine-glyoxylate aminotransferase; DAO: D-amino acid oxidase; GO: glycolate oxidase; GRHPR: glyoxylate reductase-hydroxypyruvate reductase; LDHA: lactate dehydrogenase.
Created by Biorender.com.
Ascorbic acid or vitamin C is a precursor of oxalate that is partially metabolized in the liver to dehydroascorbic acid in a reversible process. However, when ingested in excessive amounts, however, ascorbic acid is widely excreted unchanged in the urine. Another potential exogenous source is ethylene glycol, predominantly found in engine antifreeze, which is metabolized to glycolic acid, potentially causing severe hyperoxaluric crises.
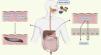
Although it is known that the body’s main source of oxalate comes from endogenous metabolism, there is a percentage that comes from the diet; this is absorbed throughout practically the entire digestive tract.12 Oxalate is an ionized conjugate base, and at the intestinal level, it tends to form complexes with divalent cations such as magnesium and calcium.13 However, free oxalate is absorbed from the lumen, mainly through paracellular passive diffusion, and is secreted through the intestinal anion transporter SLC26A6 according to the concentration gradient, functioning as a saturable receptor. Therefore, under conditions of low dietary calcium intake, oxalic acid would be soluble in the liquid part of the intestinal chyme, and in situations of alteration or saturation of the SLC26A6 channel, it would be massively absorbed into the bloodstream with a consequent reduction in loss through the gastroitestinal tract14–16 (Fig. 3).

Intestinal absorption of oxalate. Oxalate is coupled with calcium and magnesium to be eliminated with the feces. Under conditions of increased oxalate supply or low concentration of these cations, free oxalate is absorbed via the paracellular pathway and secreted again through the solute carrier family 26 member 6 (SLG26A6) transporter. The capacity to transport is limited.
Ca: calcium; Mg: magnesium; O: oxalic acid.
Created by Biorender.com.
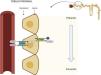
After circulating through the liver, it undergoes glomerular filtration, which also depends on its plasma concentration.15,17 Proximal tubular transport S1 and S2 is mediated by the SLC26 channel (SLC26A1), a protein located in the basolateral membrane of the cellthat captures oxalate by exchanging it for sulfate.18,19 On the apical side of the cell, the chloride-oxalate exchanger SLC26A6, reabsorbs chloride and secretes oxalate (Fig. 4). High plasma levels of oxalate will result in greater renal excretion, leading to an increased risk of precipitation with calcium in the form of calcium oxalate crystals and, consequently, kidney stones.1

Reabsorption of oxalate in the proximal tubule. In the basolateral membrane, the SLC26A1 transporter captures oxalate, and it is exchanged by sulfate. This is excreted into the tubular lumen through the SLC26A6 chloride-oxalate exchanger located on the apical surface of the cell.
SLC26A6: solute carrier family 26 member 6; Cl: chloride.
Created by Biorender.com.
Oxalate forms a soluble complex with sodium and potassium, but in the presence of calcium, it forms insoluble calcium oxalate crystals, which are predisposed by relatively low urinary pH (<7.2). Magnesium and citrate, due to their ability to bind urinary calcium, are the main inhibitors of crystallization.20
Independently of its role as an oxalate transporter, SLC26A6 could significantly modify the risk of stone formation due to its ability to interact with and inhibit the sodium/dicarboxylate cotransporter protein 1 (NaDC1) (also known as SLC13A2). Paradoxically, it has been observed that the absence of SLC26A6 in knockout mice results in an increased risk of calcium nephrolithiasis, as this leads to increased NADC1 activity and, therefore, increased reabsorption of filtered citrate and a reduced urinary citrate excretion.21
EtiologySecondary hyperoxaluria is a much more frequent cause of increased oxalate than primary hyperoxaluria, and it is produced by an increased exogenous supply of oxalate or by a deficit in its metabolism or elimination. Regarding its patophysiology, hyperoxaluria is due to three predominant etiologies.22 Firstly, increased intake of foods rich in oxalate (spinach, cabbage, beets, nuts, or tea), vitamin C (>1000 mg/day) or diets low in calcium.23 Secondly, alterations in elimination at the GI level (pancreatic insufficiency or chronic pancreatitis, celiac disease, inflammatory bowel disease or bariatric surgery).24,25 Within this subgroup, the intestinal microbiota could play a role in oxalate degradation or even in the regulation of intestinal pH,which regulates intestinal permeability.26,27 Likewise, certain foods such as octreotide or orlistat could interfere with oxalate absorption at the intestinal level.28 Thirdly, excess urinary excretion, with chronic kidney disease being the main cause, aggravates this situation due to its characteristic pro-inflammatory environment.29
Clinical presentation and diagnosisThe first step in the diagnostic approach for this entity is the determination of oxalate in urine; it is considered pathological an oxaluria greater r than 45 mg/day (>0.5 mmol/1.73 m2/day), in at least two 24-h urine samples.30 Enteric hyperoxaluria is usually present in a particularly severe form, typically with levels of 2–3 times higher than the maximum daily oxalate excretion, along with urinary calcium less than 100 mg/day and even hypocitraturia.31
Two types of calcium oxalate crystals can be found in urine: monohydrate and dihydrate, described as dumbbell and pyramid-shaped, respectively.The former is typical of primary hyperoxalurias,32 while both are can be observed in secondary forms.
Clinically, the main organ affected is the kidney, with the most common manifestations being calcium oxalate nephrolithiasis and nephrocalcinosis.19 Both entities lead to parenchymal inflammation and chronic interstitial damage, which can result in progressive renal failure and end-stage renal disease in up to 50% of cases, correlating with urine oxalate levels.9,33
Increased blood oxalate leads to crystallization, causing oxalosis, a systemic form of hyperoxaluria.9,33 Oxalate deposits may be found in various organs except the liver. One of the first clear signs of systemic oxalosis is retinal deposits.34 Bone involvement is the most disabling, since oxalate-induced osteopathy causes pain, spontaneous fractures, and is generally associated with tendinopathy and chondrocalcinosis. Diffuse demineralization and the deposition of intraosseous tophi and granulomas replace the bone marrow, which can trigger anemia resistant to erythropoietin-stimulating agents. At the cardiac level, oxalate crystals cause cardiomyopathy with alterations in cardiac conduction; at the peripheral nervous system level, they result in neuropathy; and at the vascular level, they damage the middle layer of the arteries, causing ischemic lesions, such as ulcers or even gangrene (Fig. 5).31
Histology
Calcium oxalate crystals are translucent or slightly basophilic with hematoxylin-eosin staining and their form is irregular. They are located mainly in the tubular lumen and/or within the proximal or distal tubular epithelial cells of the renal cortex, and less frequently a in the interstitium. They exhibit pronounced birefringence under polarized light, and with other complementary stains, they retain translucency.35,36
The differential diagnosis must include 2,8-dihydroxyadenine crystals, produced the in congenital deficiency of adenine phosphoribosyltransferase, in which exhibit deposits of brownish-greenish crystals with positive birefringence are observed; and also with uric acid crystals, where hematoxylin-eosin staining only identifies the empty spaces that were previously occupied by uric acid crystals, which are were generally found at the medullary level.34,35
In the renal histopathology of hyperoxaluria, fibrosis, inflammatory tubular damage, and even glomeruli without tubules can be observed.
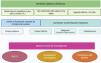
Prevention and treatmentThe therapeutic approach to secondary hyperoxaluria can be classified into three strategies: reducing the intake of oxalate and its precursors, increasing renal elimination to avoid the precipitation of oxalate crystals at the tubular level, and reducing gastrointestinal absorption of oxalate while increasing its intestinal elimination (Fig. 6).

Regarding dietary recommendations, nuts, plums, chocolate, tea, Coca-Cola, beets, and strawberries are some of the foods with high oxalate content, so reducing their consumption could be beneficial for lowering intestinal absorption. However, some studies indicate limitations in the effectiveness of this measure, since the largest source of oxalate comes from the endogenous sources. Vitamin C is a potential precursor of oxalate, so it is also recommended to avoid excessive intake, especially in patients on dialysis. A low calcium diet would increase the absorption of oxalate, so it is not recommended.1,12,37
Increasing fluid intake and using urinary crystallization inhibitors help to decrease urinary oxalate saturation.38,39 Hyperoxaluria of enteric origin is frequently associated with episodes of diarrhea, so ensuring adequate extracellular volume is especially important.19
A urinary pH below 7.2 favors the aggregation and crystallization of calcium and oxalate, so it is advisable to stabilize it between 6.2 and 6.8. Potassium citrate increases bicarbonate loss and, therefore, the alkalinization of urine, and it is one of the main inhibitors of calcium oxalate formation, so its prescription is recommended until a citraturia between 250 and 300 mg/L or 500−600 mg/day is achieved.40 In patients with renal failure, potassium citrate can be replaced wiht sodium citrate.
Calcium citrate supplements are beneficial due to their dual function41: the calcium intake provides an increase in substrate for the binding of oxalate at the intestinal level, and corrects hypocitraturia.42 Calcium-based phosphate binders are not recommended for patients with chronic kidney disease stages 3a to 5 due to the increased risk of cardiovascular mortality.43 Other non-calcium phosphate binders, such as sevelamer hydrochloride, have been investigated in an open study achieving a non-significant reduction in urinary oxalate,44; while lanthanum carbonate is currently being evaluated in a phase III trial with encouraging results.45
Magnesium supplements effectively reduce oxalate absorption due to their ability to bind to it at the intestinal level. Its main side effect is diarrhea, so its use is very limited in patients with enteric hyperoxaluria.46 Iron can be used as an alternative or complementary oxalate-binding agent, although it is less effective than calcium. Aluminum also achieves a chelating effect on oxalate, but the risk of toxicity limits its use. Special attention should be paid to the use of vitamin D analogs, as they increase intestinal absorption of calcium, decreasing the time available to bind to oxalate at the intestinal level.1
Cholestyramine is an intestinal anion exchange resin that binds cholesterol bile acids and prevents their absorption,47 increasing the availability of calcium to be bound to oxalate. Additionally, by binding intestinal oxalate helps to reduce diarrhea,48 making it a particularly useful drug in enteric hyperoxaluria.40 As side effects, it can cause hyperchloremic acidosis, as it releases chloride, and it interferes with the absorption of other medications, primarily vitamins and thiazide diuretics.
Along these lines, a 2021 quantum chemistry study suggested that trivalent cations, such as iron, aluminum, or lanthanum, as well as the element neodymium, could be attractive candidates for oxalate binding.49
The effect of the microbiome on oxalate homeostasis has been widely evaluated in animal models and in some human studies. Recently, several therapeutic formulations have been developed that explore in depth the ability of certain bacteria to degrade oxalate. The most relevant is Oxalobacter formigenes, which has demonstrated the ability to reduce urinary oxalate in rodent models of hyperoxaluria,44 inducing an increase in oxalate secretion at the intestinal level by a mechanism that is, for the moment, unknown.44
Several researchers have proposed direct supplementation with oxalate decarboxylase, an enzyme used by O. formigenes to degrade oxalate. A double-blind, randomized, controlled, crossover trial in healthy individuals showed a 24% reduction in urinary oxalate compared to placebo.50
Whole microbial transfers have been investigated as potential approaches to modulate oxalate metabolism. In rats, a fecal transplant from a herbivorous mammal (a microbiome adapted to degrade large amounts of oxalate) resulted in lower levels of oxalate in urine and feces for at least nine months compared to the ingestion of oxalate-degrading isolates.51,52 Similarly, a fecal transplant from conventional rats, which have a gut microbiome similar to that of humans, to germ-free mice reduced the amount of urinary oxalate.53 This transplant resulted in a reduction in SLC26A6 expression in the kidney and colon and an increased expression in the cecum. To date, no studies have evaluated the effect of microbiome transfer in humans.
Other species such as Bifidobacterium and Lactobacillus have also demonstrated a reduction in urinary oxalate in animals. Bacterial strains are currently under investigation in early-stage trials include Nov-001, UBLG-36, and SYNB8802. Nov-001 is a therapeutically designed microbial combination product that includes NB1000S, an oxalate-degrading bacterium, and NB2000P, a prebiotic control molecule used to regulate NB1000S abundance. A phase II trial is currently recruiting patients with enteric hyperoxaluria following an encouraging phase I study.52 UBLG-36, a strain of Lactobacillus paragasseri, is highly effective at degrading oxalate in vitro. Finally, SYNB8802, a synthetic oxalate-degrading Escherichia coli Nissle strain, administered orally, shows a significant reduction of oxalate in vitr o and in vivo; the in silico (computer) model predicts a 71% reduction in urinary oxalate in patients with enteric hyperoxaluria. Studies in mice and primates have demonstrated the ability of this molecule to consume excess intestinal oxalate, achieving a significant reduction in urinary oxalate levels in these animal models.54
The production of cytokines and several inflammatory molecules has been postulated as pathogenic agents of secondary hyperoxaluria, so immune modulation has been established as one of the possible line of research. The inhibition of the of the nucleotide-binding, leucine-rich-repeat, pyrin domain containing 3 (NLRP3) inflammasome activation could reduce the cleavage of the proinflammatory cytokines interleukin 1 beta (IL-1β) and interleukin 18 (IL-18) through caspase 1, as well as inhibit cell death induced by inflammation.55 In a study with hyperoxaluric rats, inhibition of the transient receptor potential vanilloid 1 channel mitigated reactive oxygen species-induced NLRP3 activation and calcium oxalate-induced nephropathy, although it did not reduce hyperoxaluria.56 Other NLRP3 inhibitors with promising in vivo murine results include the diarylsulfonylurea-based CP-456773, which reduces crystal-induced renal fibrosis.57
Sodium-glucose cotransporter-2 inhibitors (SGLT2i) are a revolutionary therapeutic family of antidiabetics that have demonstrated effects beyond metabolic control, reducing the risk of cardiovascular and renal events.47,49,58–60 In the field of hyperoxaluria, SGLT2i) can decrease the production of osteopontin, a glycoprotein secreted in tubular cells, which is considered one of the most important essential proteins in the matrix of calcium oxalate stones.61 It should be noted, from a mechanistic prespective, that the administration of glyoxylic acid, an oxalate precursor, to mice with sodium-glucose cotransporter-2 (SGLT2) deficiency triggered an elevated deposition of calcium oxalate crystals and an increase in the expression of osteopontin, cluster of differentiation 44 (CD44), transforming growth factor beta-1 (TGF beta-1), fibronectin 1 (Fn1) and actin alpha 2 (Acta2) compared to the control group of wild-type mice treated with the same precursor.60 In fact, a recently published Japanese observational study shows a lower prevalence of nephrolithiasis in diabetic patients treated with SGLT2i compared to other treatments.62 Therefore, the specific inhibition of SGLT2 could attenuate the formation of calcium oxalate stones in the kidney, and could become a promising therapeutic approach for this group of pathologies.
ConclusionsSecondary hyperoxaluria is a disease that is probably underdiagnosed. It is associated with different pathologies, generating a proinflammatory state that leads to a dysfunction of the SCLA26A transporter, a key factor in the pathophysiology of this entity. The increase in the prevalence of endocrine-metabolic diseases has been accompanied by a parallel rise in cases of hyperoxaluria, explained by the involvement of these diseases in its etiopathogenesis. Presently, we do not have a specific treatment, so early diagnosis and prevention are the best strategic tools.
Key concepts- -
Secondary hyperoxaluria is a pathology with an increasing incidence during recent decades and whose diagnosis is often hidden among other concomitant entities.
- -
It may result from three fundamental causes: the heightened intake of products with a high content of oxalate (such as spinach, nuts, and tea), or their precursors (such as vitamin C); increased intestinal absorption of oxalate, generally caused by a decreased absorption of fatty acids at the intestinal level (due to pancreatic insufficiency or chronic pancreatitis, celiac disease, inflammatory bowel disease or bariatric surgery); or by an increase in the renal excretion of oxalate.
- -
The rising number of cases occurs alongside the group of entities encompassed by metabolic syndrome. The chronic inflammation produces an alteration in the SLC26A6 transporter which is present both at the intestinal and renal level, and this is key in the etiopathogenesis of this disease.
- -
The main organ affected in secondary hyperoxaluria is the kidney, which may suffer damage from nephrocalcinosis or renal lithiasis, potentially progressing to end-stage renal failure. However, calcium oxalate deposits can manifest in other organs, such as the retina, heart and peripheral nervous system, with a notable absence of deposits in the liver.
- -
Treatment and prevention focus on reducing the intake of oxalate and its precursors, promoting renal elimination by avoiding the precipitation of oxalate crystals at the tubular level, and establishing targeted measures to reduce oxalate absorption and increase its intestinal elimination.



