



Receptor interacting protein kinase 3 (RIPK3) is an intracellular kinase at the crossroads of cell death and inflammation. RIPK3 contains a RIP homotypic interaction motif (RHIM) domain which allows interactions with other RHIM-containing proteins and a kinase domain that allows phosphorylation of target proteins. RIPK3 may be activated through interaction with RHIM-containing proteins such as RIPK1, TRIF and DAI (ZBP1, DLM-1) or through RHIM-independent mechanisms in an alkaline intracellular pH. RIPK3 mediates necroptosis and promotes inflammation, independently of necroptosis, through either activation of NFκB or the inflammasome. There is in vivo preclinical evidence of the contribution of RIPK3 to both acute kidney injury (AKI) and chronic kidney disease (CKD) and to the AKI-to-CKD transition derived from RIPK3 deficient mice or the use of small molecule RIPK3 inhibitors. In these studies, RIPK3 targeting decreased inflammation but kidney injury improved only in some contexts. Clinical translation of these findings has been delayed by the potential of some small molecule inhibitors of RIPK3 kinase activity to trigger apoptotic cell death by inducing conformational changes of the protein. A better understanding of the conformational changes in RIPK3 that trigger apoptosis, dual RIPK3/RIPK1 inhibitors or repurposing of multiple kinase inhibitors such as dabrafenib may facilitate clinical development of the RIPK3 inhibition concept for diverse inflammatory diseases, including kidney diseases.
La proteína quinasa 3 que interactúa con el receptor (RIPK3) es una quinasa intracelular que se encuentra a medio camino entre la muerte celular y la inflamación. La RIPK3 contiene un dominio motivo de interacción homotípica de RIP (RHIM), que permite las interacciones con otras proteínas que contienen RHIM, y un dominio de quinasa que permite la fosforilación de las proteínas diana. La RIPK3 puede ser activada a través de la interacción con las proteínas que contienen RHIM tales como RIPK1, TRIF y DAI (ZBP1, DLM-1), o a través de mecanismos independientes de RHIM en un pH intracelular alcalino. La RIPK3 media en la necroptosis y promueve la inflamación, independientemente de la necroptosis, bien a través de la activación de NFκB, o del inflamasoma. Existe evidencia preclínica in vivo de la contribución de RIPK3 a la insuficiencia renal aguda (IRA) y la enfermedad renal crónica (ERC), así como a la transición IRA-ERC derivada de ratones con deficiencia de RIPK3 o del uso de pequeñas moléculas inhibidoras de RIPK3. En dichos estudios, el tener a RIPK3 como objetivo redujo la inflamación, pero la nefropatía mejoró solo en algunos contextos. La traducción clínica de estos hallazgos se ha demorado debido al potencial de ciertas pequeñas moléculas inhibidoras de la actividad de la quinasa RIPK3 para activar la muerte celular induciendo cambios conformacionales de la proteína. Comprender mejor los cambios conformacionales de RIPK3 activadores de la apoptosis, los inhibidores duales RIPK3/RIPK1 o la reconversión de múltiples inhibidores de la quinasa tales como dabrafenib podría facilitar el desarrollo clínico del concepto de la inhibición de RIPK3 para diversas enfermedades inflamatorias, incluyendo las enfermedades renales.
- •
RIPK3 may be activated in response to diverse stimuli, including hyperosmolarity, a condition specific to the kidney under physiological conditions, as well as by ischemia-reperfusion and cytokine storms.
- •
RIPK3 activation may result in necroptosis or apoptosis and/or inflammation.
- •
In the context of kidney disease, targeting RIPK3 improved preclinical kidney disease.
- •
However, some inhibitors of the RIPK3 kinase domain induce a conformational change that promotes apoptosis, delaying clinical development of RIPK3 inhibition.
- •
A better understanding of the conformational changes in RIPK3 that trigger apoptosis, dual RIPK3/RIPK1 inhibitors or repurposing of multiple kinase inhibitors such as dabrafenib may facilitate clinical development of the RIPK3 inhibition concept for diverse inflammatory diseases, including kidney diseases.
Acute kidney injury (AKI) is defined as a sudden drop in kidney function that results in an increase in serum creatinine levels or a drop in urine output below certain thresholds.1 AKI persisting over 7 days is termed acute kidney disease.1 Chronic kidney disease (CKD) is diagnosed when kidney function is lower than a certain threshold (eGFR<60ml/min/1.73m2, i.e., grossly equivalent to the loss of one kidney) or when there is evidence of kidney injury such as albuminuria>30mg/g of urinary creatinine or other criteria for longer than three months.2 Thus, AKI and CKD are part of the same disease spectrum. CKD predisposes to AKI while AKI may cause CKD or accelerate CKD progression.3 The burden of CKD is growing fast. It is projected to become the 5th global cause of death by 2040 and the second cause of death by 2100 in some countries with long life expectancy such as Spain.4 CKD increases the risk of premature death, in part by promoting accelerated aging and systemic inflammation, dependent on the accumulation of uremic toxins and the loss of Klotho, an anti-aging and anti-inflammatory molecule of kidney origin.5 AKI is also associated with a high mortality during the acute episode, as recently evidenced during the coronavirus disease 2019 COVID-19 pandemic, as well as with increased long-term mortality.6 The current therapy of AKI and CKD is suboptimal. There is no therapy that decreases the severity or accelerates recovery from AKI while therapy for CKD slows progression but does increase kidney function.7 Cell death and inflammation are key features of AKI and CKD. Injury to kidney cells may trigger cell death and the secondary recruitment of inflammation, while systemic or local inflammation can cause AKI and CKD. Certain forms of cell death, such a regulated necrosis (e.g., ferroptosis and necroptosis) are more proinflammatory than others (e.g., apoptosis).8 Conversely, examples of systemic inflammation triggering AKI include the cytokine storm observed in sepsis and in severe COVID-19.9 We now review the role of Receptor Interacting Protein Kinase 3 (RIPK3) in kidney injury. RIPK3 is an intracellular kinase that sits at the crossroads of cell death and inflammation.
Structure and activation of RIPK3RIPK3 contains a RIP homotypic interaction motif (RHIM) domain which allows interactions with other RHIM-containing proteins and a kinase domain that catalyzes autophosphorylation and the subsequent phosphorylation of target proteins10 (Fig. 1). RIPK3 may be activated through interaction with RHIM-containing proteins such as RIPK1, TIR-domain-containing adapter-inducing interferon-β (TRIF) and DNA-dependent activator of interferon regulatory factors (DAI, also known as ZBP1 or DLM-1) or through RHIM-independent mechanisms in the presence of an alkaline intracellular pH.
 activation, and DAI/ZBP1 activated by z-RNA from viral infection. The interaction of the RHIM domain of RIPK3 with RHIM-containing proteins allows autophosphorylation and activation of RIPK3. Non-canonical RIPK3 activation is independent of the RHIM domain, and it is mediated by osmotic stress-induced intracellular acidification through NHE1.")
Different pathways of RIPK3 activation. Canonical activation of RIPK3 is dependent on its RHIM domain that binds to other RHIM-containing proteins: RIPK1 following activation by TNF receptor superfamily proteins, TRIF in response to Toll like receptor (TLR) activation, and DAI/ZBP1 activated by z-RNA from viral infection. The interaction of the RHIM domain of RIPK3 with RHIM-containing proteins allows autophosphorylation and activation of RIPK3. Non-canonical RIPK3 activation is independent of the RHIM domain, and it is mediated by osmotic stress-induced intracellular acidification through NHE1.
RIPK3 mediates both necroptosis and inflammation which in some cases is independent of necroptosis. Such functions contribute to the defense against pathogens but may also cause acute and chronic tissue injury (Fig. 2).

Functions of RIPK3. The best-known function of RIPK3 is to mediate necroptosis through phosphorylation and activation of MLKL. Necroptosis is a proinflammatory and immunogenic cell death. RIPK3 can also induce inflammation independently of necroptosis through the NLRP3-inflammsome or NFκB, although other mediators may be involved. Finally, RIPK3 has also been implicated in tissue fibrosis, although further studies are needed to discern whether RIPK3-induced fibrosis is a primary result of RIPK3 activation or inflammation-dependent. Different functions of RIPK3 contribute to tissue injury and defense against pathogens.
RIPK3 plays a key role in the execution of necroptosis. Indeed, the Nomenclature Committee on Cell Death (NCCD) defined in 2018 necroptosis as a type of regulated cell death (RCD) triggered by perturbations of extracellular or intracellular homeostasis that depends on RIPK3 activation and its downstream effector MLKL.11 Autophosphorylated RIPK3 recruits and phosphorylates mixed lineage kinase domain-like (MLKL),10,12,13 which then oligomerizes and translocates to intracellular and plasma membranes, causing membrane rupture.12
Tumor necrosis factor (TNF) receptor super family members promote binding of RIPK1 to RIPK3, which results in the assembly of different context-specific protein complexes. Complex I is activated upon TNFRSF stimulation and subsequent recruitment of TNFR-associated death domain adaptor protein (TRADD) and RIPK1, among others, and results in NF-κB activation.14 Linear ubiquitination of RIPK1 serves as a central checkpoint to regulate NFκB activation, cell proliferation and cell death.15 Under certain conditions, deubiquitylation of RIPK1 induces the formation of cytosolic complex IIa, which includes pro-caspase 8, and triggers apoptosis.14 In this context, caspase inhibitors, as seen for certain viruses, lead to the formation of the necrosome or complex IIb through the interaction of RIPK1 and RIPK3, thus inducing necroptosis.16 TNFSF members TNFα, TWEAK, FasL and TRAIL promote RIPK1/RIPK3-dependent necroptosis that contributes to various conditions, including kidney diseases.17–20
RIPK3 may also be activated by TRIF in response to Toll like receptors (TLRs) activation by stimuli such as polyinosinepolycytidylic acid (poly(I:C) and lipopolysaccharide (LPS).21 TLR4-induced necroptosis has been observed in experimental colitis and in nervous system cells.22–24
RIPK3 activation by viral RNA-DAI or cellular RNA-DAI also triggers necroptosis.14,21 When viral proteins inhibit caspases and prevent the clearance by apoptosis of virus-infected cells, DAI activation of necroptosis eliminates infected cells.25 The key role of RIPK3 in this host defense function was demonstrated in preclinical herpes simplex virus type 1 (HSV-1), influenza virus, or vaccinia virus infections.26–28
Cytosol alkalinization induced by activation of the Na+/H+ exchanger SLC9A1 also triggers RIPK3-dependent necroptosis through a non-canonical pathway, which is independent of RHIM domain, but still requires MLKL phosphorylation.29,30 Since the only described trigger of this pathway is a sudden increase in osmolarity to levels only found in the mammalian kidney medulla, this pathway would initially be expected to have little relevance in vivo, except, potentially, for kidney disease.31 In this regard, its role in kidney disease should be explored. Additionally, its potential role in any condition characterized by increased intracellular pH, independently of extracellular osmolarity, should be studied.
RIPK3 and inflammationCells dying by necroptosis release their intracellular contents promoting an inflammatory response and potentially, an immunogenic response. However, RIPK3 also triggers inflammation independent of necroptosis through activation of NFκB or the inflammasome.32
Inhibition of RIPK3 with GSK’872 reduced NLRP3 activation and inflammation in murine LPS-induced acute lung injury and in rat spinal cord injury, supporting the proinflammatory role of RIPK3.33,34 In preclinical cecal ligation and puncture (CLP)-induced sepsis, in which inflammation promotes tissue injury, RIPK3-KO mice survived longer, and had milder systemic inflammation and decreased lung, liver, and kidney injury.35–37 NFκB activation by RIPK3 is cell-type specific. RIPK3 deficient (RIPK3-KO) mouse embryonic fibroblasts (MEFs) and bone marrow derived macrophages (BMDMs) display normal NFκB activation and cytokine expression following LPS stimulation, while RIPK3 was required for NFκB activation and cytokine expression in LPS-stimulated bone marrow dendritic cells (BMDCs) and in TNFα-stimulated vascular smooth muscle cells (VSMCs).38–40 RIPK3-induced activation of NFκB was also described in pancreatic β-cells under metabolic stress where it mediates IL-1β expression, in cadmium-stimulated RAW macrophages where RIPK3 promotes M1 polarization and during osteoclastogenesis where it contributes to ovariectomy-induced bone loss in mice.41–43 The RIPK1 inhibitor necrostatin-1 prevented RIPK3-induced NFκB activation and inflammation in some experimental systems,41,43 but the precise role of RIPK1 in RIPK3-induced NFκB activation requires further in-depth characterization. In this regard, RIPK3-mediated inflammation in experimental shock induced by high dose (25μg) TNFα was independent of RIPK1, as RIPK3-KO mice were protected but necrostatin-1 accelerated death.44 However, necrostatin-1 was as protective as RIPK3 deficiency from low dose (5 or 9μg) TNFα, suggesting that RIPK1 may contribute to mild but not to severe TNF-induced shock, while RIPK3 is involved in both.35
RIPK3 also activates the NLRP3-inflammasome independently of cell death but the molecular pathway is unclear.45–48 The role of caspase-8 in RIPK3-induced activation of NLRP3 is controversial.
RIPK3 activated caspase-8 and pro-IL-1β processing in BMDCs. Conversely, caspase-8 may negatively regulate RIPK3-induced inflammasome activation, as caspase-8 deficiency increased the RIPK3-induced inflammasome activation in LPS-stimulated BMDCs. Furthermore, caspase-8 deficiency in myeloid cells increased RIPK3-induced inflammasome activation in preclinical autoimmune encephalomyelitis.45,47
However, in BMDMs, RIPK3 activated NLRP3 independently of MLKL in presence of caspase-8, while it activated NLRP3 through MLKL in caspase-8 deficient cells.46,48 Thus, the interaction between caspase-8 and RIPK3 is complex.
In humans, plasma RIPK3 levels were independently associated with acute respiratory distress syndrome among critical patients with sepsis or trauma, and they were also increased in patients exposed to mechanical ventilation and in those with moderate or severe COVID-19.49–52 However, the cellular source of plasma RIPK3 remains unclear, as does the mechanism of RIPK3 release from cells: release from living cells, membrane disruption of dying cells or contained in exosomes. It also remains unclear to what extent plasma RIPK3 may guide RIPK3-targeted therapies in the clinic.
Nevertheless, RIPK3 is critical for host defense against Streptococcus pneumoniae infection, since RIPK3-KO mice presented reduced bacterial clearance and severe lung inflammation.53 RIPK3 binding to RIPK1 and MLKL promoted mitochondrial ROS, NLRP3 inflammasome activation and necroptosis in macrophages during Streptococcus pneumoniae infection.53
Additionally, RIPK3 has been associated with fibrosis in heart, liver, and muscle. The profibrotic response induced by high glucose or the air pollutant PM2.5 in cultured cardiomyocytes is prevented by GSK‘872.54,55 In preclinical liver fibrosis induced by CCl4 or bile duct ligation, RIPK3 deficiency in macrophages reduced fibrosis and inflammation.56 Similarly, in murine Duchenne muscular dystrophy, RIPK3 deficiency reduced myofiber degeneration, inflammatory infiltrates, and muscle fibrosis.57 Since RIPK3-induced fibrosis usually occurs in parallel to an inflammatory response, further studies should discern whether RIPK3-promoted fibrosis is a consequence of the proinflammatory role of RIPK3, an independent effect, or both.
RIPK3 and PANoptosisPANoptosis refers to a pathway of inflammatory regulated cell death that activates simultaneously apoptosis, the NLRP3/inflammasome/pyroptosis, and necroptosis. PANoptosis is activated by bacterial, viral, or fungal infection in BMDMs, and by cytokines (e.g., TNFα/IFNgamma) in colon tumor cells and in BMDMs, thus it may play a key role during infection, and in the immunotherapy of cancer.58–62 PANoptosis is triggered by a protein complex called PANoptosome composed of apoptosis, pyroptosis and necroptosis proteins, including RIPK3. However, further studies are needed to fully characterize the molecular mechanisms of PANoptosis and PANoptosome assembly and to better understand its relevance in health and disease for different cell types.
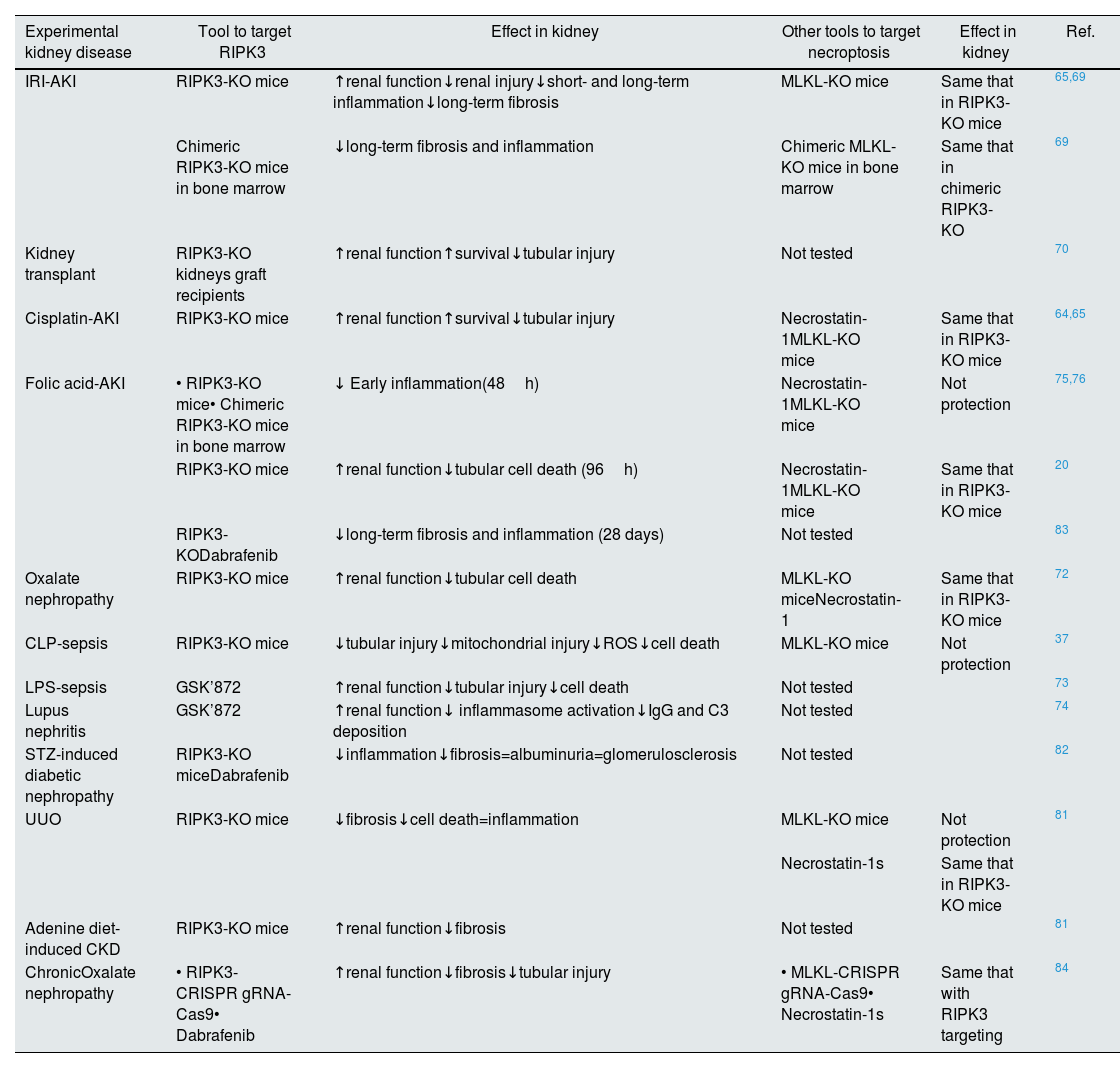
RIPK3 function in kidney diseasesIn kidney disease, there is evidence for a role of RIPK3 in necroptosis, inflammation, fibrosis, and the AKI-to-CKD transition, since targeting RIPK3 improved preclinical kidney disease (Table 1) (Fig. 3). The role of RIPK3 in kidney disease may be necroptosis-dependent or necroptosis-independent.
Targeting of RIPK3 in experimental models of kidney diseases.
| Experimental kidney disease | Tool to target RIPK3 | Effect in kidney | Other tools to target necroptosis | Effect in kidney | Ref. |
|---|---|---|---|---|---|
| IRI-AKI | RIPK3-KO mice | ↑renal function↓renal injury↓short- and long-term inflammation↓long-term fibrosis | MLKL-KO mice | Same that in RIPK3-KO mice | 65,69 |
| Chimeric RIPK3-KO mice in bone marrow | ↓long-term fibrosis and inflammation | Chimeric MLKL-KO mice in bone marrow | Same that in chimeric RIPK3-KO | 69 | |
| Kidney transplant | RIPK3-KO kidneys graft recipients | ↑renal function↑survival↓tubular injury | Not tested | 70 | |
| Cisplatin-AKI | RIPK3-KO mice | ↑renal function↑survival↓tubular injury | Necrostatin-1MLKL-KO mice | Same that in RIPK3-KO mice | 64,65 |
| Folic acid-AKI | • RIPK3-KO mice• Chimeric RIPK3-KO mice in bone marrow | ↓ Early inflammation(48h) | Necrostatin-1MLKL-KO mice | Not protection | 75,76 |
| RIPK3-KO mice | ↑renal function↓tubular cell death (96h) | Necrostatin-1MLKL-KO mice | Same that in RIPK3-KO mice | 20 | |
| RIPK3-KODabrafenib | ↓long-term fibrosis and inflammation (28 days) | Not tested | 83 | ||
| Oxalate nephropathy | RIPK3-KO mice | ↑renal function↓tubular cell death | MLKL-KO miceNecrostatin-1 | Same that in RIPK3-KO mice | 72 |
| CLP-sepsis | RIPK3-KO mice | ↓tubular injury↓mitochondrial injury↓ROS↓cell death | MLKL-KO mice | Not protection | 37 |
| LPS-sepsis | GSK’872 | ↑renal function↓tubular injury↓cell death | Not tested | 73 | |
| Lupus nephritis | GSK’872 | ↑renal function↓ inflammasome activation↓IgG and C3 deposition | Not tested | 74 | |
| STZ-induced diabetic nephropathy | RIPK3-KO miceDabrafenib | ↓inflammation↓fibrosis=albuminuria=glomerulosclerosis | Not tested | 82 | |
| UUO | RIPK3-KO mice | ↓fibrosis↓cell death=inflammation | MLKL-KO mice | Not protection | 81 |
| Necrostatin-1s | Same that in RIPK3-KO mice | ||||
| Adenine diet-induced CKD | RIPK3-KO mice | ↑renal function↓fibrosis | Not tested | 81 | |
| ChronicOxalate nephropathy | • RIPK3-CRISPR gRNA-Cas9• Dabrafenib | ↑renal function↓fibrosis↓tubular injury | • MLKL-CRISPR gRNA-Cas9• Necrostatin-1s | Same that with RIPK3 targeting | 84 |
Abbreviation: AKI: acute kidney injury; IRI: ischemia–reperfusion injury; CLP: cecal ligation and puncture; LPS: lipopolysaccharide; STZ: streptozotocin; UUO: unilateral ureteral obstruction; CKD: chronic kidney disease.
 Tubular RIPK1/RIPK3/MLKL-dependent necroptosis contributed to early renal injury in IRI-AKI and promotes an inflammatory response. RIPK3 from inflammatory infiltrates amplified kidney injury, favoring later inflammation and fibrosis. (B) In FA-AKI, ferroptosis mediated the first wave of tubular cell death leading to expression of inflammatory mediators which activated a second wave of cell death by RIPK1/RIPK3/MLKL-dependent necroptosis. In parallel RIPK3 promotes an inflammatory response at early time points, independent of necroptosis. At longer follow-up, RIPK3 also promoted inflammation and fibrosis, but it is unknown whether the chronic pro-fibrotic effect of RIPK3 is dependent or not on the initial acute injury. (C) RIPK3 promotes renal injury in inflammatory models of kidney disease such as CLP-sepsis, LPS-sepsis, and lupus nephritis. In CLP-sepsis, RIPK3-induced renal injury is mediated by NOX4 which induced mitochondrial disfunction, while in LPS-sepsis the molecular mediators of RIPK3-induced AKI are poorly characterized. In lupus nephritis RIPK3 promoted MLKL phosphorylation and inflammasome activation. (D) RIPK1/RIPK3/MLKL-dependent necroptosis contributes to oxalate nephropathy and cisplatin-AKI. (E) RIPK3 plays a role in different models of CKD and kidney fibrosis. RIPK3 promoted kidney inflammation and fibrosis in STZ-DN through inflammasome activation, and kidney fibrosis in adenine-CKD through unidentified pathways. In UUO-induced kidney fibrosis, TGFβ1 activated RIPK3, which recruited the AKT/ACL pathway to promote a profibrotic response. In chronic oxalate nephropathy, TGFβ1 mediated kidney injury through the RIPK1/RIPK3/MLKL necrosome which promoted mtROS which in turn activated CaMKII and consequently Smad2/3. Abbreviation: AKI: acute kidney injury; IRI: ischemia–reperfusion injury; FA: folic acid; CLP: cecal ligation and puncture; LPS: lipopolysaccharide; STZ-DN: streptozotocin-induced diabetic nephropathy; UUO: unilateral ureteral obstruction; CKD: chronic kidney disease; mtROS: mitochondrial ROS production.")
RIPK3 function in kidney diseases. (A) Tubular RIPK1/RIPK3/MLKL-dependent necroptosis contributed to early renal injury in IRI-AKI and promotes an inflammatory response. RIPK3 from inflammatory infiltrates amplified kidney injury, favoring later inflammation and fibrosis. (B) In FA-AKI, ferroptosis mediated the first wave of tubular cell death leading to expression of inflammatory mediators which activated a second wave of cell death by RIPK1/RIPK3/MLKL-dependent necroptosis. In parallel RIPK3 promotes an inflammatory response at early time points, independent of necroptosis. At longer follow-up, RIPK3 also promoted inflammation and fibrosis, but it is unknown whether the chronic pro-fibrotic effect of RIPK3 is dependent or not on the initial acute injury. (C) RIPK3 promotes renal injury in inflammatory models of kidney disease such as CLP-sepsis, LPS-sepsis, and lupus nephritis. In CLP-sepsis, RIPK3-induced renal injury is mediated by NOX4 which induced mitochondrial disfunction, while in LPS-sepsis the molecular mediators of RIPK3-induced AKI are poorly characterized. In lupus nephritis RIPK3 promoted MLKL phosphorylation and inflammasome activation. (D) RIPK1/RIPK3/MLKL-dependent necroptosis contributes to oxalate nephropathy and cisplatin-AKI. (E) RIPK3 plays a role in different models of CKD and kidney fibrosis. RIPK3 promoted kidney inflammation and fibrosis in STZ-DN through inflammasome activation, and kidney fibrosis in adenine-CKD through unidentified pathways. In UUO-induced kidney fibrosis, TGFβ1 activated RIPK3, which recruited the AKT/ACL pathway to promote a profibrotic response. In chronic oxalate nephropathy, TGFβ1 mediated kidney injury through the RIPK1/RIPK3/MLKL necrosome which promoted mtROS which in turn activated CaMKII and consequently Smad2/3. Abbreviation: AKI: acute kidney injury; IRI: ischemia–reperfusion injury; FA: folic acid; CLP: cecal ligation and puncture; LPS: lipopolysaccharide; STZ-DN: streptozotocin-induced diabetic nephropathy; UUO: unilateral ureteral obstruction; CKD: chronic kidney disease; mtROS: mitochondrial ROS production.
The first evidence for the role of necroptosis in AKI was the observation that the RIPK1 inhibitor necrostatin-1 protected from preclinical AKI induced by ischemia/reperfusion injury (IRI-AKI), cisplatin nephrotoxicity (Cisplatin-AKI), contrast-induced AKI (CAKI), oxalate nephropathy, and folic acid nephropathy (FA-AKI).20,63–66 The contribution of RIPK3 to necroptosis in IRI-AKI was demonstrated in RIPK3-KO mice, which were protected, even though other pathways of regulated necrosis, such as ferroptosis and mitochondrial permeability transition-regulated necrosis (MPT-RN), also contribute to IRI-AKI.65,67–69 Kidney protection appeared to depend on RIPK3 expression by kidney cells since RIPK3-KO kidney graft recipients were protected from IRI-AKI associated to the procedure.70 In this regard, freshly isolated renal tubules from RIPK3-KO mice were protected from hypoxia/reoxygenation-induced cell death.65
Necrostatin-1 and genetic deficiency of RIPK3 or MLKL improved renal function, reduced tubular injury, and increased survival in murine cisplatin-AKI.64,65 As in IRI-AKI, additional cell death pathways contribute to cisplatin-AKI since the survival of RIPK3-deficient mice increased when zVAD inhibited caspases or when caspase-8 was also deficient, suggesting that combined necroptosis and apoptosis blockade could increase protection.65 In this regard, despite kidney TUNEL-positive cells not being reduced in RIPK3-KO mice, cultured RIPK3 deficient primary tubular cells were protected from cisplatin-induced cell death.64,65
In murine FA-AKI there is a sequential activation of ferroptosis and necroptosis; an initial ferroptosis wave of cell death promotes an inflammatory response, the upregulation of RIPK3 and MLKL, and a second wave of inflammation-dependent necroptotic cell death. Indeed, RIPK3-KO mice displayed improved renal function, reduced tubular cell death, and reduced translocation of MLKL to plasma membranes in the late FA-AKI stages.20 Moreover, TWEAK, which contributes to FA-AKI, induced RIPK3-dependent necroptosis in an inflammatory milieu in cultured tubular cells, thus identifying one of the inflammatory mediators of the second necroptotic wave in FA-AKI.20
RIPK1, RIPK3 and MLKL are also overexpressed in murine oxalate nephropathy, and Necrostatin-1 or RIPK3 or MLKL deficiency reduced tubular injury and tubular cell death. MPT-RN is also involved in oxalate nephropathy, and dual inhibition of both necroptosis and MPT-RN increased protection.71 In cultured tubular cells, crystals triggered RIPK3-dependent necroptosis and MPT-RN.72
These data suggest that RIPK3 is a potential therapeutic target for different forms of AKI and in kidney transplantation, although in some cases the dual blockade of RIPK3 and other cell forms of death pathways, such as apoptosis or MPT-RN, will be more effective.
RIPK3 and inflammation in kidney diseaseThe precise role of RIPK3-mediated inflammation on overall kidney injury and dysfunction appears to be context-dependent. Thus, in AKI driven by systemic inflammation, targeting RIPK3-induced inflammation is kidney protective, but this is not the case for the early stages of ferroptosis-driven kidney injury.
RIPK3 has a pro-inflammatory role in the kidney independent of necroptosis. The main evidence that RIPK3 can mediate kidney inflammation derives from inflammatory models such as CLP-induced sepsis or LPS-induced endotoxemia, in which RIPK3 targeting was protective. Specifically, RIPK3 deficiency preserved kidney function and reduced tissue injury in CLP-induced sepsis, while MLKL deficiency was not protective, suggesting that kidney protection was not dependent on inhibit necroptosis.35,37 In CLP-induced sepsis, RIPK3 mediated kidney injury through NOX4 overexpression, production of mitochondrial ROS, mitochondrial disfunction, and cell death. However inflammatory hallmarks such as cytokine expression or inflammatory cell infiltration were not evaluated.37 In LPS-induced endotoxemia (i.e., sterile cytokine storm) GSK’872 improved renal function and reduced tubular injury and cell death.73 In human tubular cells stimulated with LPS, GSK’872 or RIPK3 downregulation by siRNA reduced cell death and apoptosis markers, but necroptotic cell death was not analyzed.73 In line with these results, patients with sepsis had higher urinary RIPK3 levels than non-sepsis patients, and sepsis patients with AKI had higher urinary and plasma RIPK3 than patients without AKI.37 These results suggest that RIPK3 may contribute to kidney injury during cytokine storms, but further studies are needed to discern the precise mechanisms involved as well as the potential role of urinary or plasma RIPK3 as a biomarker that contributes to clinical decision-making.
RIPK3 also play a role in lupus nephritis. RIPK3 inhibition with GSK’872 in NZM2328 or MLR/lpr mice reduced kidney phosphorylation of MLKL and RIPK3, inflammasome activation and IgG and C3 deposition, and improved kidney function, however as cell death was not assessed, it is unknown whether RIPK3-mediated injury in lupus nephritis is dependent on cell death or, as suggested by the reduced immune deposits, it is dependent on reduced immune cell activity.74
In murine FA-AKI, RIPK3 deficiency reduced the early inflammatory response without improving kidney function and this effect was independent of necroptosis since Necrostatin-1 or MLKL deficiency did not modify this early inflammatory response.75,76 As discussed above, ferroptosis was the primary driver of kidney injury in the first 24h of FA-AKI75 while early stages of endotoxemic or sepsis AKI are driven by systemic inflammation. The precise mechanism by which RIPK3 mediates inflammation in the early stages of FA-AKI is poorly characterized. It is dependent on RIPK3 actions in bone marrow cells, since RIPK3 is overexpressed by infiltrating inflammatory cells such as macrophages and lymphocytes in FA-AKI and bone marrow RIPK3 deficiency decreased kidney inflammation.76 NFκB activation is a prime candidate mediator, since RIPK3 deficiency in FA-AKI decreased interstitial and tubular nuclear translocation of NFκB p65, and NFκB is a well-known mediator of kidney inflammation.15,76,77 Consistent with this hypothesis, RIPK3-deficient mice had lower expression of NLRP3 and cleaved caspase 1 in FA-AKI, but NLRP3-deficient mice were not protected, ruling out a major role of the inflammasome.76
RIPK3 also modulates TWEAK-induced kidney inflammation, as this was milder in RIPK3-deficient mice.76 TWEAK is a pro-inflammatory cytokine that activates kidney NFκB without inducing cell death78 and illustrates the complex role of RIPK3 in promoting inflammation.76 RIPK3 deficiency decreased the TWEAK-induced NFκB-dependent inflammatory responses such as MCP-1, IL-1β and IL-6 expression in bone marrow derived cells, while it only inhibited the NFκB-independent production of IL-6 in tubular cells.76 However, there is a RIPK3-dependent crosstalk between both cell types, as conditioned medium from TWEAK-stimulated BMDMs activated tubular cells in a RIPK3-dependent manner.76
RIPK3, kidney fibrosis and the AKI-to-CKD transitionKidney fibrosis plays a key role in the AKI-to-CKD transition and in CKD progression.79,80 RIPK3 also contributes to kidney fibrosis in preclinical AKI-to-CKD transition and CKD.69,81–83
In IRI-AKI, RIPK3-deficient mice are protected from both early stage (2 and 7 days) necroptosis and from later (14 days, 1 month and 3 months) kidney inflammation and fibrosis.69 MLKL-KO mice displayed similar protection, suggesting that protection depends on interference with necroptosis. The initial stages of IRI-AKI depended on kidney expression of RIPK3, while RIPK3 from bone marrow-derived cells contributed to later stage fibrosis, tubular injury and inflammasome activation. In this regard, combined oxygen-glucose, and fetal bovine serum deprivation (OGFD), as might be expected in IRI, caused RIPK3-dependent necroptosis in cultured tubular cells, resulting in release of cellular contents that enhanced inflammasome activation in macrophages.69 Thus, RIPK3 from both tubular cells and bone marrow-derived cells contribute to CKD resulting from IRI-AKI through a loop of necroinflammation, in which initial RIPK3-dependent tubular cell necroptosis recruits RIPK3-dependent inflammatory responses from bone marrow cells.69
FA-AKI also causes AKI-to-CKD transition and kidney fibrosis is evident at 28 days. RIPK3-KO mice displayed less inflammation and fibrosis at this time-point. However, since RIPK3 also contributes to tubular cell death and/or inflammation in the acute phase (24–96h) of FA-AKI,20,75 it remains unclear whether the long-term protection derived exclusively from a milder initial FA-AKI or whether RIPK3 also actively participates in molecular events driving fibrosis in the chronic phase. To clarify this, RIPK3 should be targeted therapeutically, rather than prophylactically, i.e., once kidney injury is already established, for example by starting RIPK3 targeting 72–96h after induction of FA-AKI or IRI-AKI.
Kidney RIPK3 expression was also upregulated in other models of CKD, such as streptozotocin induced-diabetic nephropathy, ureteral obstruction-induced kidney fibrosis and adenine diet-induced CKD, and in human diabetic nephropathy or kidney fibrosis.81,82 In diabetic mice, RIPK3 deficiency decreased tubulointerstitial inflammation, TGF-β1 expression and fibrosis, but did not decrease glomerular injury as assessed by albuminuria or glomerulosclerosis.82 RIPK3 deficiency also improved kidney function and decreased fibrosis in murine adenine diet-CKD, but the mechanism was not studied.81 RIPK3 deficiency also decreased unilateral ureteral obstruction (UUO)-induced kidney fibrosis in mice but had no impact on kidney inflammation or TGF-β1 expression,81 suggesting the presence of shared and specific targets for diverse cause of CKD. RIPK3-induced fibrosis following UUO likely involved AKT-dependent activation of ATP citrate lyase (ACL), since kidney phospho-AKT and phospho-ACL were reduced in RIPK3-deficient mice. Indeed, in cultured fibroblasts, RIPK3 regulated fibrogenesis downstream of TGF-β1 through activation of the AKT-ACL pathway.81 The profibrotic effect of RIPK3 in UUO was independent of MLKL, as MLKL-KO mice were not protected, and may be dependent on RIPK1, as necrostatin-1 prevented AKT-ACL activation and reduced fibrotic gene expression in cultured fibroblasts and necrostatin-1s reduced fibrosis and tubular injury in UUO kidneys.81,84 In chronic oxalate nephropathy, intrarenal injection of CRISPR gRNAs for either RIPK3 or MLKL suppressed fibrosis and leukocyte infiltrates and improved renal function.84 In fact, in chronic oxalate nephropathy TGFβ1 induced fibrosis and leukocyte recruitment through RIPK1/RIPK3/MLKL necrosome formation, mitochondrial ROS production, calcium/calmodulin-dependent protein kinases-II (CaMKII) and Smad2/3.84
Therapeutic options to interfere with RIPK3 in kidney diseasesAlthough several RIPK3 inhibitors have shown promising preclinical results, no RIPK3 inhibitor is under clinical development for inflammatory diseases85 (https://clinicaltrials.gov/, accessed August 1, 2022). Dabrafenib and GSK’872 have been tested in preclinical kidney disease. Dabrafenib is a B-Raf inhibitor in clinical use for malignancy, that is also a type I ATP-competitive RIPK3 inhibitor.
Dabrafenib inhibited necroptosis in tubular cells exposed to crystals or crystalline particles.86 In vivo, dabrafenib increased survival and reduced fibrosis in murine streptozotocin-induced diabetic nephropathy, and reduced inflammation, and fibrosis in FA-AKI-to-CKD transition, in chronic oxalate nephropathy and in UUO-induced kidney fibrosis.82–84 However, dabrafenib has not been tested in forms of AKI where necroptosis plays a key role, and these studies are needed. Additionally, the combination of dabrafenib with the MEK inhibitor trametinib has been associated with nephrotoxicity in postmarking studies.87 Other BRAF inhibitors also inhibit RIPK3 kinase activity, including AZ628, regorafenib, PLX4720, vemurafenib, and sorafenib.85 There is scarce information on these drugs and kidney disease. However, vemurafenib is nephrotoxic due to off-target ferrochelatase inhibition.88 Sorafenib protects from UUO-induced experimental kidney fibrosis, but whether this is related to Ripk3 inhibition has not been assessed.89,90
GSK’872 inhibits RIPK3 kinase activity by binding to the kinase domain. It protected mice from lupus nephritis, FA-AKI-to-CKD transition, and streptozotocin-induced diabetes kidney disease.74,82,83 Additionally, GSK’872 prevented TWEAK/TNFα/IFNγ-induced necroptosis in tubular cells and TWEAK-induced inflammation in tubular and Jurkat cells.20,76 However, GSK‘872 is marred by inducing a RIPK3 kinase-dead conformation that increases the interaction with RIPK1, enhances RIPK1/FADD/CASP8 complex formation and initiates RIPK1 kinase-dependent apoptotic cell death.91 This issue is shared by other RIPK3 inhibitors such as HS-1371 and GSK’843 and has decreased the impetus for clinical evaluation of RIPK3 inhibitors.85,92 However, inhibition of RIPK3 kinase activity by dabrafenib does not appear to favor apoptosis.85 Inhibitors 9 and 18 are highly selective for RIPK3 over RIPK1 and RIPK2, but still insufficiently characterized.93 A further RIPK3 inhibitor, Zharp-99, blocked necroptosis and protected against TNF-induced systemic inflammatory response syndrome in mice.94 Finally, GSK’067 and GSK’074 target both RIPK1 and RIPK3 and block necroptosis without inducing apoptosis.85
Summary and future perspectivesIn summary, despite expanding in vivo preclinical evidence of the contribution of RIPK3 to AKI, CKD, and the AKI-to-CKD transition through induction of necroptosis and/or inflammation, clinical translation has been delayed by the potential of some small molecule RIPK3 inhibitors to trigger apoptotic cell death. A better understanding of the conformational changes in RIPK3 that trigger apoptosis, dual RIPK3/RIPK1 inhibitors or repurposing of multiple kinase inhibitors such as dabrafenib may facilitate clinical development of the RIPK3 inhibition concept for diverse inflammatory diseases, including kidney diseases. In this regard, the recent availability of a comprehensive database of the structure of almost all human proteins may contribute to advance the field of RIPK3 inhibition (https://alphafold.ebi.ac.uk/; accessed August 1, 2022).
Sources of supportFIS/Fondos FEDER (PI19/00588, PI19/00815, PI22/00469, PI22/00050, DTS18/00032), ERA-PerMed-JTC2018 (KIDNEY ATTACK AC18/00064 and PERSTIGAN AC18/00071, ISCIII-RETIC REDinREN RD016/0009), Sociedad Española de Nefrología, Sociedad Madrileña de Nefrología (SOMANE), FRIAT, Comunidad de Madrid en Biomedicina B2017/BMD-3686 CIFRA2-CM. Instituto de Salud Carlos III (ISCIII) RICORS program to RICORS2040 (RD21/0005/0001) funded by European Union – NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR) and SPACKDc PMP21/00109, FEDER funds. Salary support: AC18/00064 to MF-B, ISCIII PFIS to AML-D, Ramon y Cajal program (RYC2019-026916-I) to ABS, FPU program (FPU18/01341) to JG-M.
Conflict of interestAO has received grants from Sanofi and consultancy or speaker fees or travel support from Advicciene, Astellas, Astrazeneca, Amicus, Amgen, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Mundipharma, Kyowa Kirin, Alexion, Freeline, Idorsia, Chiesi, Otsuka, Novo-Nordisk, Sysmex and Vifor Fresenius Medical Care Renal Pharma and is Director of the Catedra Mundipharma-UAM of diabetic kidney disease and the Catedra Astrazeneca-UAM of chronic kidney disease and electrolytes. The rest of authors have nothing to disclose.



