


Cell death is a finely regulated process occurring through different pathways. Regulated cell death, either through apoptosis or regulated necrosis offers the possibility of therapeutic intervention. Necroptosis and ferroptosis are among the best studied forms of regulated necrosis in the context of kidney disease. We now review the current evidence supporting a role for ferroptosis in kidney disease and the implications of this knowledge for the design of novel therapeutic strategies. Ferroptosis is defined functionally, as a cell modality characterized by peroxidation of certain lipids, constitutively suppressed by GPX4 and inhibited by iron chelators and lipophilic antioxidants. There is functional evidence of the involvement of ferroptosis in diverse forms of kidneys disease. In a well characterized nephrotoxic acute kidney injury model, ferroptosis caused an initial wave of death, triggering an inflammatory response that in turn promoted necroptotic cell death that perpetuated kidney dysfunction. This suggests that ferroptosis inhibitors may be explored as prophylactic agents in clinical nephrotoxicity or ischemia–reperfusion injury such as during kidney transplantation. Transplantation offers the unique opportunity of using anti-ferroptosis agent ex vivo, thus avoiding bioavailability and in vivo pharmacokinetics and pharmacodynamics issues.
La muerte celular es un proceso minuciosamente regulado que se desarrolla a través de diferentes vías. La muerte celular regulada, ya sea mediante apoptosis o necrosis regulada, ofrece la posibilidad de introducir una intervención terapéutica. La necroptosis y la ferroptosis se encuentran entre las formas mejor estudiadas de necrosis regulada en el contexto de la nefropatía. Revisamos los datos actuales que avalan que la ferroptosis desempeña una función en la nefropatía y las repercusiones que tiene este conocimiento en el diseño de nuevas estrategias terapéuticas. La ferroptosis se define de forma funcional como una modalidad celular caracterizada por la peroxidación de ciertos lípidos, constitutivamente suprimida por GPX4 e inhibida por quelantes férricos y antioxidantes lipofílicos. Existen datos probatorios funcionales de la implicación de la ferroptosis en diversas formas de nefropatía. En un modelo de lesión renal aguda nefrotóxica bien caracterizado, la ferroptosis provocó una ola inicial de muerte, la cual desencadenó una respuesta inflamatoria que a su vez promovió la muerte celular necroptótica que perpetuó la disfunción renal. Esto sugiere que los inhibidores de la ferroptosis pueden explorarse como agentes profilácticos en la nefrotoxicidad clínica o en la lesión por isquemia-reperfusión, como durante un trasplante de riñón. Los trasplantes ofrecen una oportunidad única para el uso de agentes inhibidores de la ferroptosis ex vivo, con lo que se evitarían los problemas de biodisponibilidad y los problemas de farmacocinética y farmacodinámica in vivo.
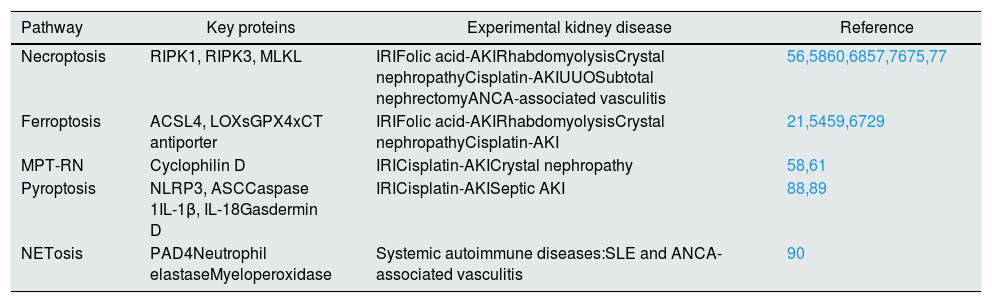
Cell death is a finely regulated process occurring through different molecular pathways. Whereas apoptosis is an active and programmed cell death mode, necrosis has been classically defined as an accidental cell death not subjected to molecular regulation. However, in recent years, different forms of regulated necrosis have been described, including necroptosis, ferroptosis, pyroptosis, or mitochondria permeability transition regulated necrosis (MPT-RN)1,2 (Table 1). A common feature of regulated necrosis is cell membrane rupture with release of intracellular contents promoting an inflammatory response that may amplify tissue injury and activate other cell death modalities. This auto-amplification loop driven by necrosis has been named necroinflammation.3
Mechanism of regulated necrosis and relevance for kidney disease.
| Pathway | Key proteins | Experimental kidney disease | Reference |
|---|---|---|---|
| Necroptosis | RIPK1, RIPK3, MLKL | IRIFolic acid-AKIRhabdomyolysisCrystal nephropathyCisplatin-AKIUUOSubtotal nephrectomyANCA-associated vasculitis | 56,5860,6857,7675,77 |
| Ferroptosis | ACSL4, LOXsGPX4xCT antiporter | IRIFolic acid-AKIRhabdomyolysisCrystal nephropathyCisplatin-AKI | 21,5459,6729 |
| MPT-RN | Cyclophilin D | IRICisplatin-AKICrystal nephropathy | 58,61 |
| Pyroptosis | NLRP3, ASCCaspase 1IL-1β, IL-18Gasdermin D | IRICisplatin-AKISeptic AKI | 88,89 |
| NETosis | PAD4Neutrophil elastaseMyeloperoxidase | Systemic autoimmune diseases:SLE and ANCA-associated vasculitis | 90 |
IRI: ischemia–reperfusion injury; UUO: unilateral ureteral obstruction; SLE: systemic lupus erythematosus; ANCA: antineutrophil cytoplasm antibody.
The best-characterized form of regulated necrosis is necroptosis,4 defined as a necrotic cell death dependent on the kinase activity of RIPK1, which, upon stimulation, phosphorylates and activates RIPK3, which in turn activates MLKL, forming a complex that induces plasma membrane rupture.5
In ferroptosis, cell death occurs through massive lipid peroxidation that leads to plasma membrane rupture.1 Glutamate-induced cell death mediated by inhibition of the cystine/glutamate antiporter Xc and consequent oxidative stress was termed “oxytosis” in the early 2000s.6 However, oxytosis and ferroptosis are now considered two names for the same cell death pathway.7,8
MPT-RN is triggered by perturbations of the intracellular microenvironment, such as elevated calcium concentrations, which induce an abrupt loss of the impermeability of the inner mitochondrial membrane to small solutes following opening of a so-called mitochondrial permeability transition pore (mPT) which requires Cyclophilin D. This results in rapid Δψm dissipation, osmotic breakdown of both mitochondrial membranes, and necrotic cell death.1 In some circumstances, mPT can trigger apoptosis due to cytochrome c release.5
Pyroptosis is a non-apoptotic caspase-dependent cell death which ultimately drives the cleavage of gasdermin D, which translocates to the plasma membrane and binds phospholipids, thus inducing the formation of pores that ultimately cause membrane lysis.9,10
Extracellular traps (ETs) are web-like structures composed of double stranded DNA, histones, antimicrobial peptides, and proteases, ejected by immune cells to ensnare microbes in a sticky matrix of extracellular chromatin and microbicidal proteins.11 ETs were first observed in neutrophils and called NETs (neutrophil ETs). Netotic cell death or NETosis consists of the release of granular enzymes to the nucleus, followed by histone citrullination, chromatin decondensation, nuclear envelope disruption, release of NETs and cell death.12 However, under some conditions, neutrophils releasing NETs remain viable.13,14 This is usually associated with release of mitochondrial DNA, rather than nuclear DNA.12 RIPK3/MLKL-dependent lytic death may also result in NET generation.15 Macrophages may also release ETs, called METs and undergo METosis.16
Molecular mechanisms of ferroptosisFerroptosis is a caspase-independent form of cell death characterized by massive lipid peroxidation at the plasma membrane and/or subcellular compartments such as mitochondria, endoplasmic reticulum or lysosomes7,17–19 (Fig. 1). Ferroptosis is involved in neurotoxicity, neurodegenerative disease, liver and heart ischemia/reperfusion injury (IRI) and acute kidney injury (AKI).20,21 Sensitivity to ferroptosis depends on different biological processes, including amino acid, iron, and polyunsaturated fatty acid metabolism, and the biosynthesis of glutathione, phospholipids, NADPH, and coenzyme Q10.22 Reduced glutathione (GSH) is the major intracellular antioxidant and glutathione biosynthesis is limited by cysteine availability. In this sense, inhibition of the cystine/glutamate antiporter Xc promotes cell death by ferroptosis in some cell types. Xc− is inhibited directly by erastin or its more potent analogs imidazole ketone erastin (IKE) and piperazine erastin (PE), or indirectly by sorafenib or high extracellular glutamate concentrations.7,23 However, some cells use the transsulfuration pathway to synthesize cysteine becoming resistant to Xc− inhibitors.24 Moreover, buthionine-(S, R)-sulfoximine (BSO), an inhibitor glutamate-cysteine ligase, interferes with GSH synthesis and can also promote ferroptosis.25
 increases phosphatidylethanolamine-AA species which can be oxidized by lipoxygenase (LOX) leading to cell death by ferroptosis. ACSL4 and LPCAT3 favor ferroptosis since they mediate AA-PE species generation. In contrast, glutathione peroxidase 4 (GPX4) reduces lipid hydroperoxides (L-OOH) producing oxidized glutathione (GSSG) and negatively regulating ferroptosis. Inhibitors of the Xc antiporter, of glutathione synthesis and of GPX4 activity are inductor of ferroptosis since they reduce cellular anti-oxidative capacity. Ferroptosis can be prevented by scavengers of lipid peroxidation, inhibitors of phospholipid synthesis and LOXs, iron chelators and lipophilic antioxidants.")
Molecular mechanisms of ferroptosis. Exacerbated esterification of arachidonic acid (AA) increases phosphatidylethanolamine-AA species which can be oxidized by lipoxygenase (LOX) leading to cell death by ferroptosis. ACSL4 and LPCAT3 favor ferroptosis since they mediate AA-PE species generation. In contrast, glutathione peroxidase 4 (GPX4) reduces lipid hydroperoxides (L-OOH) producing oxidized glutathione (GSSG) and negatively regulating ferroptosis. Inhibitors of the Xc antiporter, of glutathione synthesis and of GPX4 activity are inductor of ferroptosis since they reduce cellular anti-oxidative capacity. Ferroptosis can be prevented by scavengers of lipid peroxidation, inhibitors of phospholipid synthesis and LOXs, iron chelators and lipophilic antioxidants.
Additionally, loss of glutathione peroxidase 4 (GPX4) activity elicits ferroptosis. GPX4 is a GSH-dependent enzyme that reduces lipid hydroperoxides (L-OOH) into the corresponding alcohols (L-OH) producing oxidized glutathion (GSSG). Genetic knockdown of GPX4 or its chemical inhibition with RSL3 increased intracellular lipid hydroperoxide levels and promoted ferroptotic cell death independently of Xc− status.26 FINO2 and FIN56 were recently identified as compounds that decrease GPX4 activity either directly or indirectly.27
Lipid metabolism is a key driver of cell sensitivity to ferroptosis: the amount and localization of polyunsaturated fatty acids (PUFAs) determine the occurrence of ferroptosis. Phospholipid derivatives of arachidonic acid (AA) or adrenoyl acid are most susceptible to lipid peroxidation during ferroptosis.28–30 In this line, Acyl-CoA synthetase long-chain family member 4 (ASCL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), both involved in the insertion of PUFAs, such AA, into cell membranes, have been identified as potential points of regulation of ferroptosis. In fact, AA containing phosphatidylethanolamine (AA-PE) species is specifically oxidized during ferroptosis and incubation with AA promotes ACSL4-dependent ferroptosis in cultured fibroblasts.30,31 Moreover, ACSL4 contributes to ferroptosis in breast cancer cells and in intestinal IRI.31,32. However, LPCAT3 is not required for ferroptosis induction.31
Both, non-enzymatic (Fenton chemistry) and enzymatic (lipoxygenases, LOXs) mechanisms may trigger lipid peroxidation.33,34 However, the specific contribution of each mechanism is unclear and it may differ for different ferroptosis inducers.34 At which regulatory point iron contributes to ferroptosis is unclear. Iron is required for Fenton Chemistry, but LOXs are non-heme iron-containing proteins whose function could be regulated by iron levels.35 Further studies are necessary to determine the relative contribution of iron to both pathways.
The molecular link between lipid peroxidation and cell death remains unclear. Potential mechanisms include membrane disruption, formation of protein pores, and generation of toxic oxidation products as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE).33
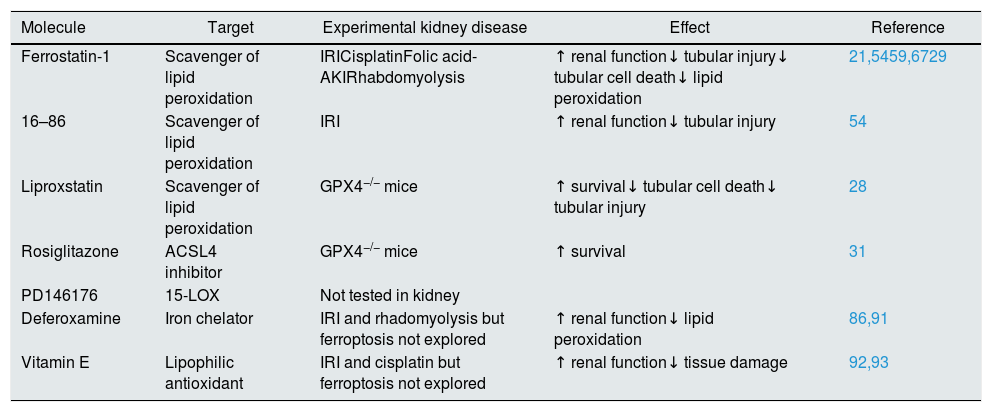
From a therapeutic point of view, pharmacological inhibitors of ferroptosis may be used to prevent ferroptosis-induced tissue injury. These include inhibitors of ACSL4 or LOXs, iron chelators, lipophilic antioxidants, and the small molecules ferrostatins and liproxstatins that inhibit lipid peroxidation, possibly by acting as radical-trapping antioxidants22 (Table 2). All these inhibitors have been broadly used to assess the role and molecular regulation of ferroptosis in cultured cells, but their use in vivo is limited by low stability and efficacy. Thus, the development of more stable ferroptosis inhibitors that are safe to use in human disease is an unmet clinical need.
Targeting ferroptosis in experimental AKI.
| Molecule | Target | Experimental kidney disease | Effect | Reference |
|---|---|---|---|---|
| Ferrostatin-1 | Scavenger of lipid peroxidation | IRICisplatinFolic acid-AKIRhabdomyolysis | ↑ renal function↓ tubular injury↓ tubular cell death↓ lipid peroxidation | 21,5459,6729 |
| 16–86 | Scavenger of lipid peroxidation | IRI | ↑ renal function↓ tubular injury | 54 |
| Liproxstatin | Scavenger of lipid peroxidation | GPX4−/− mice | ↑ survival↓ tubular cell death↓ tubular injury | 28 |
| Rosiglitazone | ACSL4 inhibitor | GPX4−/− mice | ↑ survival | 31 |
| PD146176 | 15-LOX | Not tested in kidney | ||
| Deferoxamine | Iron chelator | IRI and rhadomyolysis but ferroptosis not explored | ↑ renal function↓ lipid peroxidation | 86,91 |
| Vitamin E | Lipophilic antioxidant | IRI and cisplatin but ferroptosis not explored | ↑ renal function↓ tissue damage | 92,93 |
IRI: ischemia reperfusion injury; LOX: lipoxygenase.
Recently, ferroptosis has been reported as a relevant cell death subroutine in cardiomyocytes. Myocardial ferroptosis promotes heart failure (HF), myocardial infarction, heart transplantation and doxorubicin-induced cardiotoxicity.22 Cardiac IRI is a critical event leading to HIF-induced transferrin receptor-1 upregulation and iron overload, reactive oxygen species generation, lipid oxidative damage and ferroptosis.36 Moreover, mitochondrial iron accumulates in rat myocardial IRI cell-permeable iron chelators decreased myocardial injury.37 Iron accumulation also induced Nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated Heme-oxygenase (Hmox) up-regulation, mitochondrial lipid peroxidation and ferroptosis in doxorubicin and IRI-induced HF.38 Myocardial Toll like receptor 4 (TLR4) and NADPH oxidase 4 (NOX4) know-down also reduced ventricular remodeling and cardiomyocyte ferroptosis in rat HF.39
Ferroptosis contribution to chronic obstructive pulmonary disease pathogenesis was demonstrated in either GPX4 deficient or overexpressing mice.40
A role of ferroptosis in hereditary hemochromatosis progressing to liver cirrhosis was hypothesized. Liver accumulation results from loss-of-function mutations in genes encoding negative regulators of iron absorption. Indeed, in murine hereditary hemochromatosis, ferrostatin prevented liver damage as judged by lower levels of lipid peroxidation, serum transaminase activity and liver collagen deposition.41 The most common liver disease, simple steatosis during nonalcoholic fatty liver disease, may led to steatohepatitis characterized by lipid droplet accumulation, inflammation and fibrosis. Similar to the role of ferroptosis as the initial driver of cell death folic acid-induced AKI, discussed below, ferroptosis is the main driver, rather than apoptosis and necroptosis, of steatohepatitis in a mice fed a choline-deficient, methionine-supplemented diet.42
There are several in vivo links between ferroptosis-related events and human or experimental neurological disorders (e.g. Alzheimer, Parkinson, ferroptosis-related events, and Huntington disease, amyotrophic lateral sclerosis, Friedreich ataxia, traumatic brain injury, stroke, periventricular leukomalacia), namely iron accumulation in specific brain neuronal areas, reduced GSH activity or content, reduced GPX4 protein levels and cell loss.43 For instance, iron accumulates in areas susceptible to Alzheimer's disease that show plaque formation, even before aggregation of Aβ and tau proteins which is also favored by oxidative stress.44–46
Ferroptosis markersTherapeutic interventions benefit from the availability of biomarkers that provide information on the state of activation of the therapeutic target and monitor the effectiveness of the therapeutic intervention in suppressing the target. Thus, the hallmarks of ferroptosis are potential biomarkers in the clinic. Oxidation products such as MDA or 4-HNE could be markers of lipid peroxidation during ferroptosis. In this regard, MDA and 4-HNE levels are increased in pathological situations where ferroptosis could have a clinical relevance such as neurodegenerative and cardiovascular diseases.47–51 In this line, a clinical trial in Amyotrophic Lateral Sclerosis confirmed the predictive value for six of four biomarkers associated to ferroptosis, neuronal integrity, DNA oxidation, lipid peroxidation and iron status.52 However, additional pathways could modulate these biomarkers and their specificity for ferroptosis is highly questionable. Therefore, it would be desirable the identification of specific markers of ferroptosis. ACSL4 expression is increased in experimental kidney and intestinal IRI and this correlated with the severity of the injury.32,53 In addition, tubular ASCL4 expression was increased in human post-transplantation AKI.53 While ASCL4 activation contributes to ferroptosis, whether ACSL4 levels (not activity) provides information on ferroptosis occurrence is yet unclear. Recent reports indicate that lipid peroxidation during ferroptosis could have a specific pattern. PE-AA is most susceptible to suffer oxidation during ferroptosis.28,29 Thus, the analysis of the oxy-lipidome could provide ferroptosis biomarkers.
Ferroptosis and kidney diseaseFerroptosis has been involved in several forms of AKI by functional in vivo studies targeting ferroptosis with chemical inhibitors or gene expression modulation. Indeed, it has shown to cause synchronized necrosis of renal tubules in models of IRI and oxalate crystal-induced AKI.54 Synchronization is the well-known phenomenon of some tubules being severely affected while others are relatively preserved during kidney injury.
Kidney IRIThe kidneys are highly susceptible to IRI, which represents a common clinical problem. Kidneys are injured both during both ischemia and subsequent reperfusion, leading to tubular cell death, AKI, and potentially to subsequent a progressive chronic kidney injury. There is accumulating evidence for a role of regulated necrosis and, specifically, ferroptosis, in kidney IRI. Both ferrostatin-1 and its more stable analog 16–86 prevented AKI in mice with severe kidney IRI, reducing tubular injury and improving renal function, but 16–86 was more effective. In addition, combined inhibition of necroptosis, MPT-RN and ferroptosis offered complete protection from ultra-severe kidney IRI, being more effective that monotherapy. This suggests that the three pathways cooperate to cause AKI in kidney IRI.54 In this regard, in cell culture experiments, deficiency of the necroptotic protein MLKL favored the occurrence of ferroptosis, and absence of the pro-ferroptotic protein ACSL4 favored the occurrence of necroptosis.53 Furthermore, in kidney IRI, MLKL deficiency accelerated the increase in ACSL4 expression, potentially suggesting accelerated ferroptosis when necroptosis is suppressed, but prophylactic 16–86 followed by additional doses every 3h was not protective.53,54 Despite the authors suggestion that increased ACSL4 represents increased ferroptosis when necroptosis is suppressed, the fact that 16–86 failed to prevent kidney dysfunction in MLKL deficient mice indeed argues against recruitment of ferroptosis in this model, and, consequently argues against a role for ACSL4 as a marker of ferroptosis in vivo.
Toxic and crystal-induced AKINephrotoxicity and crystal-induced kidney injury are also frequent causes of AKI. Experimental cisplatin and folic acid nephropathy are frequently studied models which have a human counterpart.55 Folic acid-AKI is characterized by a tubular cell death and there is evidence that early injury is mediated by ferroptosis: whereas single prophylactic doses of either or both apoptosis or necroptosis inhibitors were not protective, a single prophylactic dose of ferrostatin-1 reduced renal injury and improved renal function.21 Moreover, ferrostatin-1 prevented the inflammatory response and the expression of necroptotic proteins, suggesting that ferroptosis may activate other pathways of cell death, including necroptosis, which was previously shown to depend on the proinflammatory cytokine TWEAK and its receptor Fn14 in this model.21,56 In this regard, folic acid nephropathy could represent an excellent model to study ferroptosis biomarkers, given the demonstrated role of ferroptosis, the high levels of kidney 4-HNE and oxygenated AA-PE species and the response of both markers to ferrostatin-1.21,29 Moreover, in folic acid nephropathy, ferroptosis is an early event that triggers an inflammatory response and a secondary wave of necroptotic cell death that amplifies and maintains kidney dysfunction56 (Fig. 2).

Interplay of different forms of regulated necrosis in the course of AKI and therapeutic implications. Ferroptosis is an early event that triggers an inflammatory response and, dependent of inflammation, a secondary wave of necroptotic cell death that amplifies and maintains kidney dysfunction. Ferroptosis occurs before current diagnosis of AKI and its targeting could be useful for prevention, while, necroptosis is sensitive to therapeutic intervention since is initiated after AKI has been triggered.
Cisplatin is a nephrotoxic cancer chemotherapy drug. Genetic targeting or chemical inhibition of necroptosis preserved renal function in cisplatin nephrotoxicity in vivo and decreased tubular cell death in vitro.57,58 Ferroptosis also contributes to cisplatin nephrotoxicity. In mice, a single prophylactic dose of ferrostatin-1 attenuated kidney injury and improved renal function assessed 3 days later. The enzyme myo-inositol oxygenase, a proximal tubular enzyme that exacerbates cellular redox injury, played a key role in cisplatin-induced ferroptosis.59 However, discrepant results were reported in cultured tubular cells. Ferrostatin-1 weakly decreased cisplatin-induced HK2 death, while the necroptosis inhibitor necrostatin-1 was not protective.59 HK-2 cells are a human proximal tubular cell line. By contrast, blocking necroptosis protected primary murine tubular cells from cisplatin-induced death.57 Different experimental conditions may account for this discrepancy: different sources of tubular cells and different timing and doses of cisplatin.57,59 Blocking necroptosis and ferroptosis under the same experimental conditions may solve the discrepancy.
Crystal nephropathies may trigger three different pathways of regulated necrosis: necroptosis, ferroptosis and MPT-RN. The role of both necroptosis and MPT-RN has been studied in-depth, and prophylactic inhibition of both pathways offered protection at 24h after injury.60,61 In addition, the protection offered by prophylactic ferrostatin-1 supports a contribution of ferroptosis. Ferrostatin-1 pretreatment preserved renal function, and decreases tubular injury and inflammation in a murine model of crystal nephropathy.54 Dual necroptosis and MPT-RN blockade was more effective than monotherapy, but ferrostatin-1 combinations were not tested.61
Heme proteins (hemoglobin and myoglobin) are endogenous nephrotoxic agents released from cells upon intravascular hemolysis or rhabdomyolysis, respectively. Both are freely filtered by glomeruli, and cause tubular cell oxidative stress, inflammation, mitochondria dysfunction, cell death that may be apoptotic in nature, and AKI.62–66 Ferroptosis also contributes to rhabdomyolysis-associated AKI.67 Although there was evidence of activation of apoptosis and necroptosis pathways in the kidneys during rhabdomyolysis, only ferrostatin-1 reduced oxidative stress, cell death and preserved kidney function, both in vivo and in vitro, whereas no beneficial effects were observed after administration of the pan-caspase inhibitor zVAD or in RIPK3 deficient mice.68
Ferroptosis plays a key role in hemoglobin-induced cell death in several cell types. For instance, hemin overload increased oxidative stress, platelet activation and monocyte death, an effect partially prevented by ferrostatin-1.69,70 Similarly, ferrostatin-1 reduced neuron lipid peroxidation and cell death after hemorrhagic stroke.71 However, whether ferroptosis mediates hemoglobin-mediated kidney disease is still unclear. Necrostatin-1 reduced hemin-induced mouse cortical collecting duct cell death while ferrostatin-1 was not protective.72 However, proximal tubular cells may represent a more appropriate model and in vivo confirmation is needed.
Chronic kidney disease (CKD)The potential contribution of ferroptosis to chronic tissue injury was conclusively demonstrated in doxorubicin-induced cardiac injury and fibrosis.38 Interestingly, doxorubicin, a well-known inducer of podocyte injury,73 also increased renal Ptgs2 mRNA, a putative marker of ferroptosis.38 Ferroptosis may also contribute to chronic progressive tissue injury during infection. Thus, Pseudomonas aeruginosa lipoxygenase (pLoxA) oxidized host AA-PE to 15-hydroperoxy-AA-PE (15-HOO-AA-PE), and triggered ferroptosis in human bronchial epithelial cells.74 However, evidence that regulated necrosis or regulated necrosis-associated molecules contribute to CKD progression relates mainly to necroptosis. Thus, the necroptosis inhibitor necrostatin-1 preserved renal function in rats with subtotal nephrectomy75 and decreased kidney inflammation and fibrosis in unilateral ureteral obstruction76 while targeting necroptosis in myeloid cells reduced NET formation and glomerular injury in experimental ANCA vasculitis.77 Since synchronized regulated necrosis may promote nephron loss and contribute to the AKI-to-CKD transition and it is triggered by necroptosis and executed by ferroptosis, a combination therapy employing necrostatins and ferrostatins was hypothesized to protect from irreversible nephron loss.78 However, AKI findings should not be directly extrapolated to long-term preservation of renal function and necroptosis targeting findings may not be directly related to putative ferroptosis targeting findings. While necrostatin-1 aggravated experimental polycystic kidney disease probably by decreasing the loss of cyst wall cells,79 ferroptosis was hypothesized to be protective, although the evidence, so far, is indirect. Thus, cyst growth depends on transepithelial chloride secretion, through the chloride channels cystic fibrosis transmembrane conductance regulator (CFTR) and TMEM16A (anoctamin 1). Plasma membrane phospholipid peroxidation activated TMEM16A, and this activation was inhibited by ferrostatin-1 and other compounds.80 In any case, the putative protective roles of ferroptosis in CKD should be demonstrated in in vivo ferroptosis targeting studies.
Evidence from human kidney diseaseIn humans, tubular cell death was the best histopathological correlate of renal dysfunction in the so-called acute tubular necrosis form of AKI.81 Acute tubular necrosis (a term coined in the 19th century an unrelated to current concepts of cell death modalities) is the most common form of AKI. Despite initial suggestions that apoptosis was a key contributor to human AKI, no apoptosis-targeting therapy is yet in clinical use. By contrast, there is evidence that ferroptotic cell death is a feature of human AKI. Urinary casts in patients with AKI resemble morphologically to ex vivo experiments with isolated tubular segments exposed to erastin.54 Additionally, ACSL4 immunostaining was observed in proximal tubules from human kidney biopsies with AKI following kidney transplantation or in thrombotic microangiopathy, but no in normal kidneys.53 However, its potential association with severity of injury was not assessed and whether ACSL4 immunostaining actually identifies ferroptotic cells is unclear. Kidney PE-ox levels are increased in experimental folic acid-AKI, where ferroptosis plays a key role, and also the urinary sediment of AKI patients, regardless of AKI cause.29 Altogether, this information suggests that ferroptosis may occur in human kidney disease, but unraveling its precise role in the course of kidney disease awaits the performance of clinical trials.
Therapeutic options to interfere with ferroptosisPreclinical studies have showed that ferroptosis can be successfully modulated in AKI in vivo by different drugs, and in some cases the combined targeting of different regulated necrosis pathways further improved the outcomes. Therefore, approaches to be tested in clinical trials may consist on prophylactic administration of one or several compounds targeting one or several regulated necrosis pathways in high risk situations such as patients undergoing heart surgery or in preservation solutions used for kidney grafts. Table 2 summarizes agents with the potential to inhibit ferroptosis.
Among antioxidant agents, despite ferrostatin-1 successful use as a single dose prophylactic agent in Folic Acid induced AKI,21 it is very unstable in vivo. Indeed, the more stable analog ferrostatin 16–86 was more effective than Fer-1 in experimental kidney IRI.54 Another compound named SRS 11–92 was protective ex vivo in isolated renal tubular cells exposed to hypoxia/reoxygenation and iron/hydroxyquinolone.82 Also, liproxstatin-1 prevented kidney injury resulting from inducible GPX4 depletion.28 Vitamin E and its quinone/hydroquinone metabolites had anti-ferroptotic activity in mouse striatal cells stimulated with RSL3,83 and were protective in IRI and cisplatin-AKI although these studies were previous to ferroptosis description and the effect over ferroptosis was not studied. Erastin-induced ferroptosis was reduced by the antioxidant trolox.7,82
Additional drugs shown to decrease erastin-induced ferroptosis include ebselen (glutathione peroxidase mimetic) and the MEK inhibitor U0126,7 as well as baicalein which suppressed erastin-mediated GPX-4 degradation in pancreatic cancer.84
Rosiglitazone has ACSL4 inhibitor y effect and prolongs survival in GPX4−/− mice.31 While the drug is no longer available for clinical use for diabetes in Europe due to safety concerns, the short term and even single dose use needed in AKI may be safe. Alternatively, new molecules may be designed based on rosiglitazone structure.
PD146176, a 15 LOX inhibitor prevents cell death in GPX−/− mouse embryonic fibroblast and in human fibrosarcoma cells exposed to erastin.31,85
Iron chelation prevents ferroptosis and diverse iron chelating strategies were successfully used in experimental heme-associated AKI in the early nineties (e.g. 86). Retrospectively, they may have been interfering with ferroptosis. However, none of them is currently in clinical use. Dose-adjusted deferasirox may be tested. It accumulates in proximal tubular cells, driving iron depletion-induced cell death.87 Precisely because of this proximal tubular cell tropism, it may be tested at lower doses to induce enough iron depletion to prevent ferroptosis.
Summary and future perspectivesIn summary, ferroptosis is a proinflammatory form of regulated necrosis characterized by lipid peroxidation that may be targeted in vivo by small molecules and genetic manipulation. There is evidence for the occurrence of ferroptosis during AKI and its contribution to the pathogenesis of diverse forms of AKI. Indeed, ferroptosis may be an early event driving the amplification of kidney injury through recruitment of inflammation and other forms of regulated necrosis. Thus, we envision that clinical trials in humans may start by testing whether ferroptosis inhibitors protect from ischemia–reperfusion kidney injury during transplantation: this is a predictable for of AKI that allows ex vivo therapeutic maneuvers. Ex vivo perfusion of the grafts with ferroptosis inhibitors may bypass safety, pharmacokinetics and pharmacodynamics issues inherent to in vivo administration. In this regard, experimentally, the feasibility of protecting from AKI by a single prophylactic dose of ferroptosis inhibitors has been demonstrated. Unmet needs include the availability of ferroptosis inhibitors better suited for in vivo use as well as clarification of the optimal timing of ferroptosis inhibitor therapy and of the role of ferroptosis in CKD.
Conflict of interestsThe authors declare that they have no conflict of interest.
FIS/Fondos FEDER (PI15/00298, CP14/00133, PI16/02057, PI16/01900, PI18/01366), ISCIII-RETIC REDinREN RD016/0009, ERA-PerMed-JTC2018 (KIDNEY ATTACK AC18/00064 and PERSTIGAN AC18/00071), DTS18/00032, Sociedad Española de Nefrología, FRIAT, Comunidad de Madrid en Biomedicina B2017/BMD-3686 CIFRA2-CM. Salary support: ISCIII Miguel Servet and to A.B.S. and M.D.S.-N., B2017/BMD-3686 CIFRA2-CM to M.F.-B. and D.M.-S., ISCIII Sara Borrell to J.M.-M.M., Ramon y Cajal program to J.A.-M., and ISCIII PFIS to M.G.-H.



