



We describe the case of a patient with renal disease associated with a B-cell lymphoid infiltrate, with no associated systemic disease and with the presence of monoclonal IgM-kappa paraprotein.
Renal involvement is known to be associated with lymphoplasmacytic neoplasia and is not considered rare (14–34% depending on whether the biopsy is pre mortem or post mortem), but it often goes undiagnosed due to the absence of symptoms and sometimes the lack of biopsies performed in these patients.1 However, the occurrence of primary low-grade B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) is considered unusual.2 These tumours were initially described by Isaacson and Wright3 in the gastrointestinal tract, and later in the thyroids, lungs and salivary glands. More recently, B-cell MALT lymphomas have been reported in a large variety of locations, including the urogenital tract, although as mentioned, their occurrence is very rare.3 The presence of a glomerular lesion associated with lymphoplasmacytic neoplasia is not uncommon, and could be the direct result of the lymphoplasmacytic disorder through deposition of a paraprotein (amyloid) or monoclonal immunoglobulin, or its origin could be mediated by an immune mechanism that causes membranoproliferative, membranous or minimal change glomerulonephritis (GN).4 There are reported cases of isolated renal involvement with no evidence of systemic disease, haematological malignancy or associated autoimmune disease which are sometimes associated with a serum monoclonal IgG or IgM-kappa component.5
This patient was a 71-year-old man admitted because oedema. He had a 1-year history of hypertension and post-traumatic subdural haematoma drained 6 years earlier. Due to the presence of renal failure (serum creatinine 2.5mg/dL) with of nephrotic syndrome (albumin: 1.9g/dL; total cholesterol: 315mg/dL; urinary protein: 6.6g/24h) and normal-sized kidneys by ultrasound, a renal biopsy was performed. Other additional tests showed elevated beta-2 microglobulin and lactate dehydrogenase, serum protein electrophoresis and immunofixation with the presence of monoclonal IgM-kappa gammopathy (monoclonal component 0.2g/dL) and urine negative for Bence-Jones protein. During hospitalisation, the patient presented a deterioration in renal function (Cr 3.8mg/dL). Treatment was therefore initiated with oral prednisone at a dose of 1.5mg/kg/day plus diuretics (furosemide and spironolactone) and angiotensin II receptor blockers in an attempt to control the proteinuria.
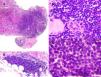
The renal biopsy (Figs. 1 and 2) revealed the presence of: (1) dense lymphoid infiltrates, immunophenotypically atypical, suggestive of a low-grade B cell lymphoproliferative process, and (2) mesangioproliferative glomerulonephritis, with few exudative changes and no demonstrable immune deposits.
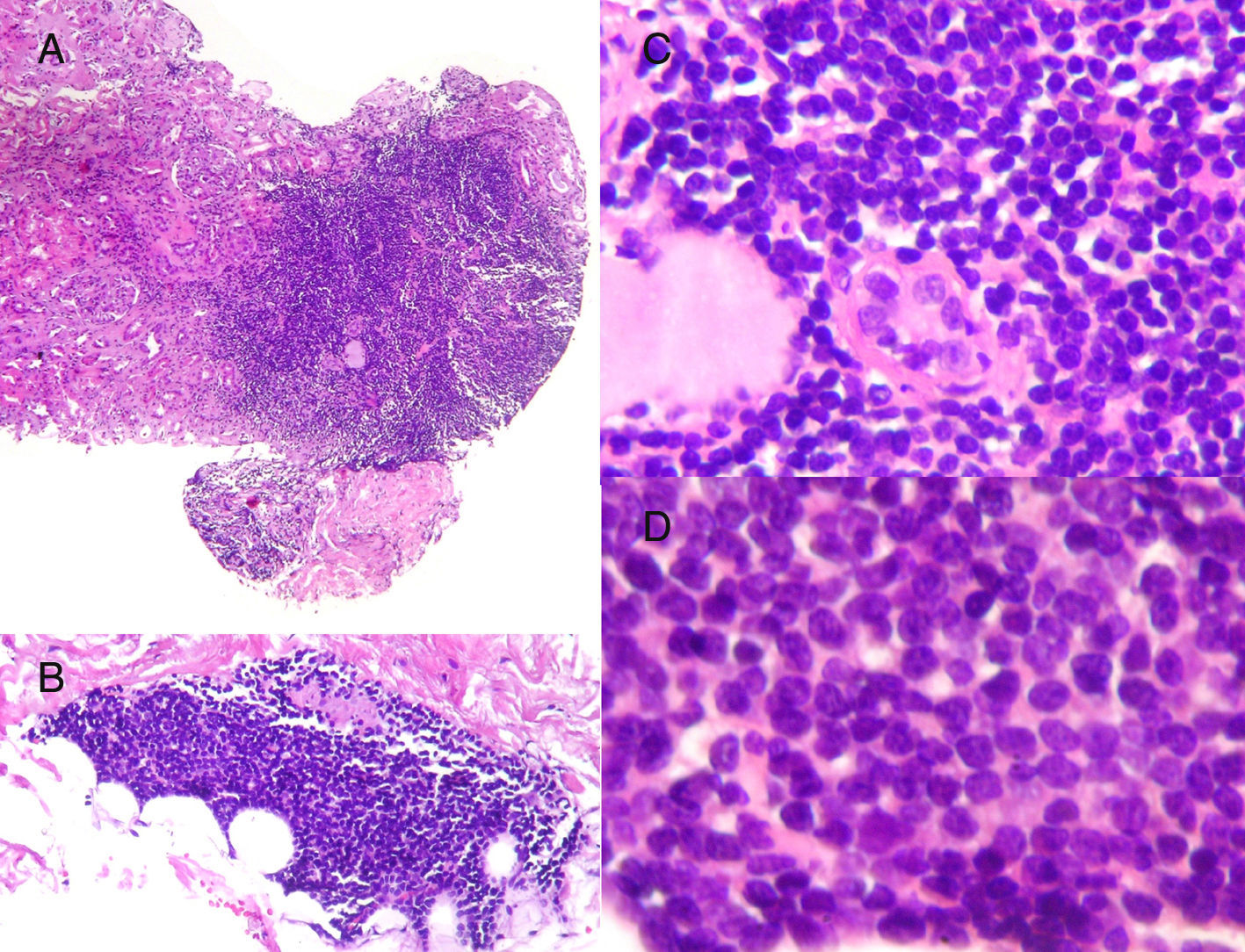
Histological characteristics of the lymphoid infiltrates that affected both the renal parenchyma (A: H&E, ×10) and the perirenal tissue (B: H&E, ×10). The lymphoid infiltrate had destroyed the parenchyma, leaving isolated residual tubules with images that may suggest lymphoepithelial lesion (C: H&E, ×20). Cells were monomorphic, small in size, with an irregular nuclear outline, barely visible nucleolus and no significant mitotic activity (D: H&E, ×40).
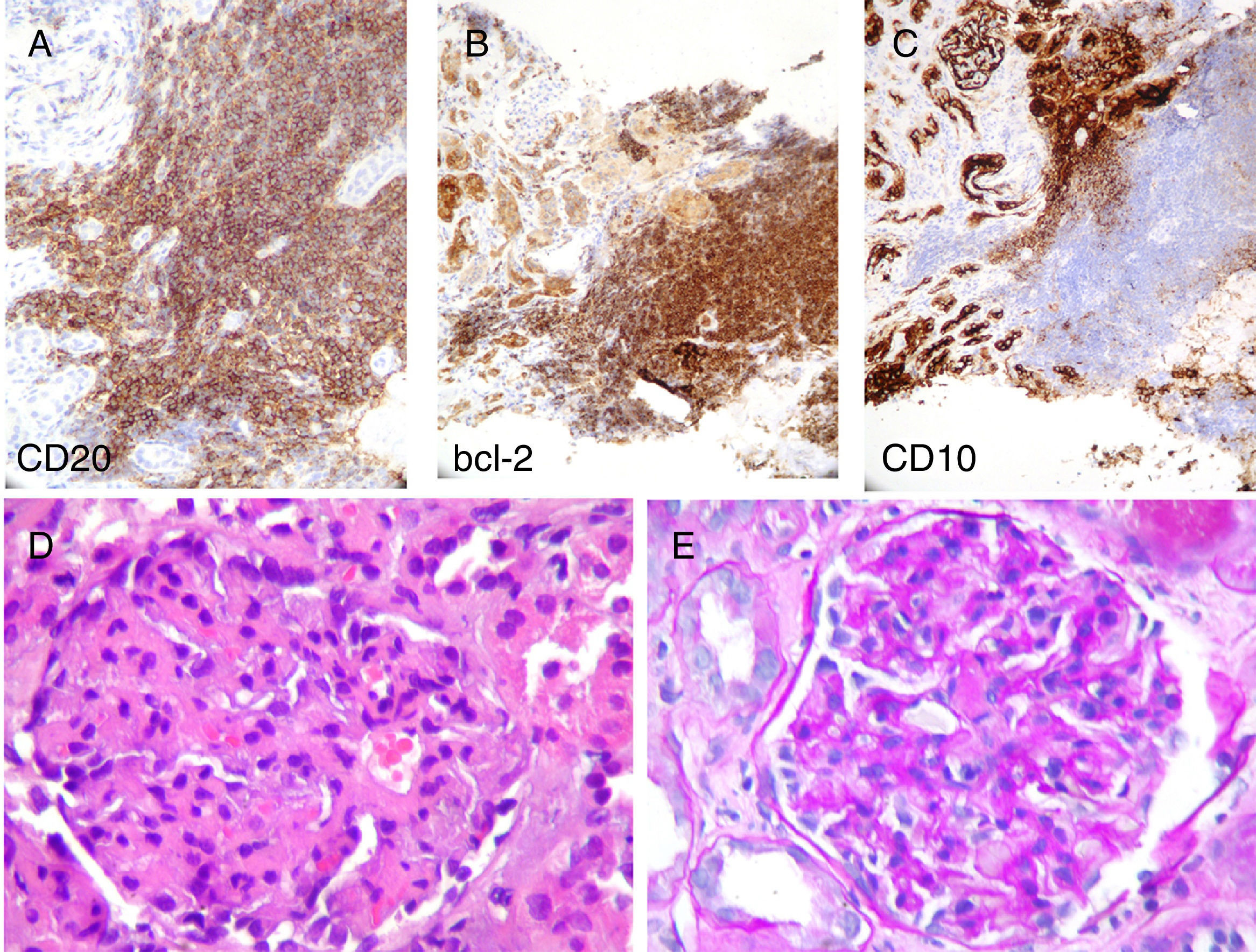
The lymphoid population expressed CD20 and bcl-2, with negative staining for CD10 and other B lymphoid lineage markers. (A–C: immunohistochemistry technique indicated in figure, ×20). In preserved normal renal parenchyma, it can see lobular glomeruli with increased matrix and, to a lesser extent some mesangial cellularity with occasional images of endocapillary proliferation (D: H&E, ×40; E: PAS-diastase, ×40).
Given the biopsy findings and the presence of monoclonal IgM-kappa gammopathy, the haematology department was consulted to assess the presence of a possible chronic lymphoproliferative process that had associated renal infiltration, as the most likely cause of the symptoms. Iliac crest biopsy was performed, with a diagnosis of mixed mature B and T cell lymphoid infiltrate, probably reactive.
The patient recovered renal function quickly with oral prednisone and diuretics, achieving normal renal function (1.1mg/dL) and a reduction of proteinuria to 260mg/day in 2 months, despite persistence of the monoclonal IgM-kappa component (0.1g/dL). After 9 months of follow-up by nephrology and haematology departments, he continues to have no evidence of progression of the lymphoproliferative process, with normal renal function and persistence of the monoclonal IgM-kappa component (0.1–0.2g/dL).
In our case, there was no evidence (clinical, analytical or imaging tests) that made us suspect renal lymphoma. The biopsy was suggestive of a low-grade B cell lymphoproliferative process together with mesangioproliferative glomerulonephritis with no demonstrable immune deposits. The literature describes the association of lymphomas with numerous forms of glomerulonephritis: membranous GN,4 membranoproliferative GN4,5 and minimal change GN.4 In many cases, like ours, the clinical presentation is determined by the renal disease associated with the lymphoproliferative process, presenting nephrotic proteinuria with or without associated renal insufficiency.5 Our patient started with nephrotic syndrome secondary to the presence of mesangioproliferative GN; therefore, the initiation of steroid treatment from the time at which we performed the renal biopsy and a favourable response to steroids resulted in the rapid recovery of renal function (Cr 1.1mg/dL) with disappearance of the proteinuria (260mg/day) in only 2 months.
Following the result of the renal biopsy, the haematology department was consulted. Having ruled out an associated chronic lymphoproliferative process, and given the absence of symptoms, we decided to adopt a watch and wait approach with close follow up of the patient's clinical progress. After 9 months, the patient remained asymptomatic, with normal renal function and no proteinuria, with persistence of the monoclonal IgM-kappa component. Although the initial diagnosis suggested a poor short-term prognosis, the absence of systemic involvement seems to have contributed to a favourable outcome.
Please cite this article as: Merino García E, Pérez del Barrio MP, Borrego Hinojosa J, Borrego Utiel FJ, Sánchez Perales MC. Infiltración renal linfomatosa en paciente con síndrome nefrótico. Nefrologia. 2018;38:92–94.



