




Severe and prolonged hypophosphatemia is exceedingly rare and may be life-threatening.1 This commentary presents a 28-year-old Caucasian woman who endorsed fatigue, muscle weakness and palpitations that started 2 weeks prior to admission. Her medical history included abnormal uterine bleeding and iron deficiency anemia. She received 2 doses of intravenous (IV) ferrous carboxymaltose (FCM, 750mcg/dose) approximately 8 weeks prior to presentation. Her physical showed decreased muscle strength in upper and lower extremities and mild tenderness in her thighs. Her serum phosphorus was found to be repeatedly low (1–1.6mg/dL) despite appropriate replacement. She had an abnormally high fractional excretion of phosphorus at 21%, and a 24-h urine excretion of phosphorus at 3.4g/day (reference: 0.3–1.3g/day). C-terminal fibroblast growth factor-23 (c-FGF23) levels were found to be within normal limits (116RU/mL, reference: 44–215RU/mL).
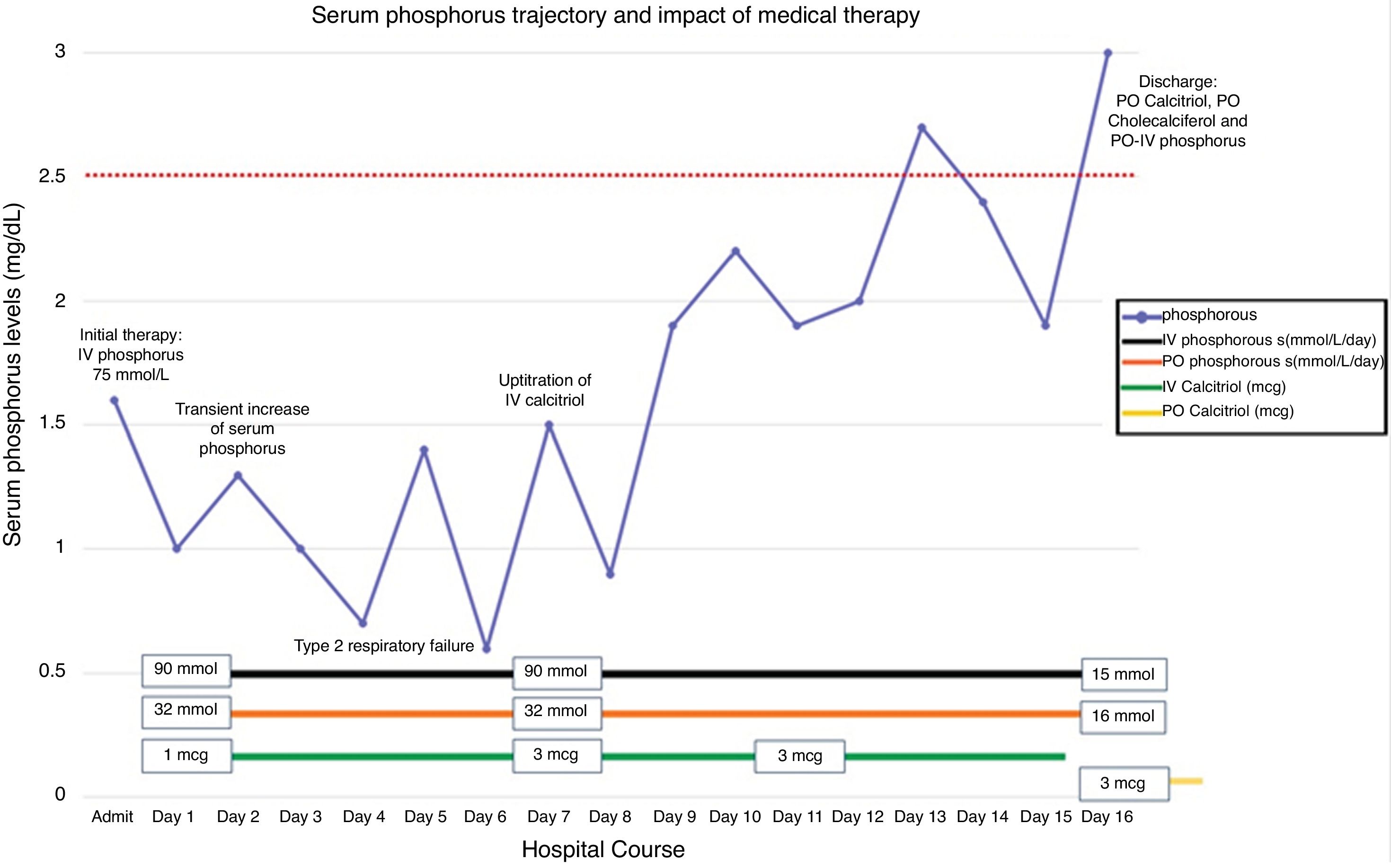
The patient was aggressively treated with both IV phosphorus (up to 90mmol/L/day) and oral phosphorus (up to 32mmol/L/Day). Intravenous calcitriol was carefully added with a starting dose of 1mcg/day. On day 7, her serum phosphorus level decreased critically as low as 0.6mg/dL and the patient developed respiratory failure. The dose of IV calcitriol was slowly up-titrated to 3 mcg/day over the following days. Two weeks later, her serum phosphorus concentration was noted to increase to borderline low levels (Fig. 1, day 14). She was discharged with oral calcitriol, cholecalciferol and phosphorus replacement. Three months later, her serum phosphorus level stabilized without needing supplementation.
Approximately, 70% of phosphate is absorbed in the small intestine especially in the proximal aspects which is facilitated by 1,25(OH)2 vitamin D3 (calcitriol) via type 2b sodium-phosphate transporters.1–2 Parathyroid hormone (PTH) may increase the absorption of calcium and phosphorus in the gut by upregulating the synthesis of calcitriol. It also can reduce phosphorus reabsorption by affecting the Na-Phosphate co-transporters 2a and 2c in the proximal nephron.1,2 Furthermore, fibroblast growth factor – 23 (FGF23) is a phosphaturic hormone synthesized by osteocytes and is an important regulator of phosphorus homeostasis.9
Recent evidence suggests that FGF23 secretion can be favored by other factors such as chronic inflammation and iron deficiency.3,4 These conditions may enhance the expression of hypoxia inducible factor (HIF) genes and upregulate the synthesis of FGF23 messenger RNA in the osteocyte. The activity of FGF23 is further regulated by post-translational processes. Its biologically active form – intact FGF23 (iFGF23) – is cleaved by putative proteases (yet to be characterized) into inactive N-terminal and C-terminal FGF23 fragments.3 The phosphaturic effects of iFGF23 are mediated through the FGF23 receptor 1 and the co-receptor klotho, which is abundantly distributed in otherwise healthy kidneys. FGF23 induces a negative phosphate balance by means of: (1) down-regulation of the activity of Na-Phosphorus co-transporters (2a and 2c), (2) down regulation of the activity of 1-α hydroxylase, which reduces the formation of calcitriol and (3) upregulation of 24-α hydroxylase, which increases the concentrations of biologically inactive forms of vitamin D.1,3,4 Also, FGF23 provides a negative feedback for the secretion of PTH through its receptors in the parathyroid cells.1
After demonstrating a renal wasting syndrome, the next step was to uncover a FGF23-dependent or FG23-independent etiology. There are two assays to evaluate FGF23 activity: (a) iFGF23 assay and (b) C-terminal FGF23 assay. The former measures c-FGF23 and iFGF23 (“total FGF23 production”) without indicating the relative amounts of each peptide. In patients with iron deficiency anemia, there is an increased synthesis of FGF23 which is offset by high protease activity. However, when intravenous FCM is infused, the effect of those proteases is blunted, and effects of iFGF23 prevail; inducing severe hypophosphatemia. Furthermore, c-FGF23 fragments tend to decrease after IV iron supplementation; potentially related to decreased degradation of FGF23.3–5 In this case, the patient's c-FGF23 was measured approximately 8 weeks after the dose of FCM; limiting an accurate assessment of the effect of FGF23. For this reason, a comprehensive clinical evaluation was conducted; see Table 1.6–10 FCM infusion was identified as the potential physiological stressor.
Renal phosphate wasting syndromes [see references 6–10].
| Condition | Genetic mutation | Comments |
|---|---|---|
| FGF23 mediated | ||
| TIO | N/A | Paraneoplastic syndrome of a mesenchymal tumor producing FGF23. Associated with prostate cancer, oat cell cancer, or hematologic malignancies. Presents with bone pain, fractures, muscle pain and weakness. |
| XLH | Activating mutation PHEX gene | XLD. More prevalent in male children. Presents with bone pain, tooth abnormalities, impaired growth and rickets of the femur and tibia. |
| ADHR | Activating mutation FGF23 gene | Extremely rare. Incomplete penetrance. Manifests with short stature, rickets of the lower extremities (children). Adults present with bone pain, repeated fractures and fatigue |
| ARHR | Inactivating mutation DMP1 or ENPP1 | Unclear epidemiology. Clinical presentation in adults is similar to ADHR. |
| Fibrous dysplasia | Activating mutation of GNAS1 | Diagnosed early in childhood. Unknown incidence and prevalence due to the number of undiagnosed cases. Non-inherited developmental anomaly. Presents with slow growing bone abnormalities, bone pain, swelling and tenderness. |
| Linear nevus sebaceous syndrome | Genetic mosaicism possibly involving KRAS | Presents in 1:10,000 patients. Benign lesion at birth which can progress to BCE in adulthood. Renal involvement can lead to rickets, skeletal abnormalities, hypophosphatemia, seizures and intellectual disabilities. |
| Iron polymaltose infusion | N/A | Acquired. Thought to be related to inactivation of proteases that naturally inactivate iFGF23 into C-fragments and F-fragments. |
| Non-FGF23 mediated | ||
| Hyperparathyroidism | N/A | High PTH levels. Vitamin D deficiency can worsen the hypophosphatemia in these patients. |
| HHRH | Mutations in SLC34A3 | AR disorder. Characterized by hypophosphatemia secondary to renal phosphate wasting. Similar presentation to other phosphorus wasting syndromes. Distinguished by hypercalciuria. |
| Fanconi syndrome | Variable | Idiopathic, genetic or acquired. Clinically, patients presents with polyuria, polydipsia, dehydration, bone abnormalities, impaired growth. Presents as rickets in children and osteomalacia in adults. |
| Dent's syndrome | Mutations in CLCN5 and OCRL | XLR. Mainly affects males in childhood. Presents with proteinuria, hypercalciuria, and nephrocalcinosis. Rickets and short stature are common. Disease progression leads to kidney failure by early to mid-adulthood. |
| Drug-induced | N/A | Different mechanisms. Potential drugs: mannitol, tetracyclines, toluene, ifosfamide, cisplatin, aminoglycosides, anti-retroviral medications, imatinib, bisphosphonates, diuretics, etc. |
ADHR: autosomal dominant hypophosphatemic rickets, AR: autosomal recessive disorder, ARHR: autosomal recessive hypophosphatemic rickets, BCE: basal cell epithelioma, CLCN5: chloride channel 5, DMP1: dentrin matrix protein-1, ENPP1: ectonucleotide pyrophosphatase/phosphodiesterase-1, FGF23: fibroblast growth factor 23, HHRH: hereditary hypophosphatemic rickets with hypercalciuria, OCRL: inositol polyphosphate-5-phosphatase, KRAS: KRAS proto-oncogene GTPase, SLC24A3: solute carrier family 34 member 3, PHEX: phosphate regulating endo-peptidase homolog X-linked, PTH: parathyroid hormone, TIO: tumor induced osteomalacia, XLH: X-linked hypophosphatemia, XLD: X-linked dominant, XLR: X-linked recessive.
There are no guidelines for the management of FCM-induced hypophosphatemia. Although Burosumab neutralizes FGF23 and has been shown to be effective for the treatment of X-linked hypophosphatemic rickets, this medication has not been approved in the management of cases such as this one. Although the administration IV calcitriol has been anecdotally reported in the literature, its administration is controversial.4 Calcitriol can increase FGF23 synthesis through local stimulation of vitamin D sensitive receptors (VDRs) in osteocytes3–7 while increasing the intestinal absorption of phosphorus. Therefore, it is possible that the latter effect may not be enough to compensate for the severe urinary phosphorus losses.
This case highlights the importance of a physiologically-driven approach to the diagnosis and management of hypophosphatemia. Renal wasting syndromes can be further characterized by investigating FGF23 levels and understanding its role in phosphorus balance. Iron deficiency anemia followed by FCM infusion may increase the risk of prolonged and severe forms of hypophosphatemia which may represent a therapeutic challenge. Clinicians should be aware of the effect of FCM and base their decisions on the physiological and clinical characteristics of each case.
Ethics approval and consent to participateFormal research ethics approval was not sought for this case report.
Availability of data and materialsAll data generated are included in this article.
Authors’ contributionGV drafted the manuscript. GV, AC, KJM & AM were part of the medical team responsible for the care of the patient. KJM provided expert consultation for the management of the patient and the elaboration of this manuscript. GV, AC, IP, KJM & AM critically reviewed the manuscript and provided significant intellectual contributions to it. All the authors reviewed the last version of the manuscript and approved it.
FundingNone.
Conflict of interestThe authors declared no conflicts of interest regarding to the research, authorship, and/or publication of this article.



