






Classically, aldosterone actions are associated with the stability of the effective circulating volume and with blood pressure control, while parathormone actions are linked to bone mineral metabolism, calcium, and phosphate homeostasis. Nevertheless, the relationship between these two hormonal axes surpasses these areas. A bidirectional interrelation between calcium-phosphorus metabolism and blood pressure control can lead to alterations in both. This can have significant implications for the evolution and treatment of patients. To illustrate this relationship, we present two clinical cases that demonstrate the pathophysiology involved.)
Clásicamente, asociamos la acción de la aldosterona (ALD) con la estabilidad del volumen circulante eficaz y el control de la tensión arterial (TA) y la acción de la parathormona (PTH) con el control del metabolismo óseo mineral y la normalidad de calcemia y fosforemia, sin embargo, la relación entre estos dos ejes hormonales trasciende estos ámbitos y podemos definir una interrelación bidireccional entre ambos, que determina la patogenia de las alteraciones en el metabolismo calcio- fósforo y el control de la TA, lo que implica consecuencias en la evolución y tratamiento de los pacientes. La exposición de dos casos clínicos nos permite exponer la fisiopatología de esta relación.
A 44-year-old man with Whiteveen-Kolk syndrome (15q24 microdeletion) visits the clinical center due to obesity and unstudied arterial hypertension (HTN) that had not been evaluated. Laboratory tests showed preserved renal function, hypernatremia, hypokalemia with inadequate urinary potassium excretion, normocalcemia with hypercalciuria, hypophosphatemia with hyperphosphaturia, elevated parathormone (PTH), decreased renin (R) levels with elevated aldosterone (ALD) and ALD/R ratio. Thyroid ultrasound and parathyroid (PT) and PT scan were normal. Abdominal CT scan showed bilateral adrenal hyperplasia (ARH). The patient was treated with spironolactone and potassium supplementation. The serum potassium levels became normal and the hypercalciuria subsides (Table 1).
Case 2
A 72-year-old male who has been evaluated in outpatient clinic for suspected primary hyperparathyroidism (PTH). He suffered from HTN, overweight and analytically hypercalcemia with hypercalciuria, hypophosphatemia with hyperphosphaturia and occasional elevation of PTH. Neither neck ultrasound nor sestaMIBI parathyroid scintigraphy with Tc 99m clearly identified PT adenoma. No nephrolithiasis, densitometry with femoral and lumbar osteopenia. Given the persistence of hypercalcemia and hypophosphatemia compatible with hyperparathyroidism, the ultrasound was repeated, which identified an intrathyroidal nodule, so elective nodulectomy was performed to diagnose PT adenoma. After surgery, calcemia and PTH levels were immediately normalized, but hypophosphatemia with hyperphosphaturia persisted for several months despite adequate vitamin D levels. After 6 months calcium/phosphorus metabolism normalizes, but he develops de novo hypokalemia, hyperkaliuria with metabolic alkalosis, R in normal range and ALD at high limit of normality. An abdominal CT scan identifies images of bilateral adrenal gland hyperplasia. Blood pressure (BP) normalizes and the analytical alterations disappear after initiating treatment with spironolactone (Table 1).
What we know about aldosterone and PTHIt should not be difficult to understand that the mechanisms of calcium regulation are interconnected to the mechanisms responsible for maintaining an adequate extracellular volume. The most efficient way to decrease the excess of extracellular calcium is to increase calciuria. An increase in calciuria occurs when hypercalcemia is detected by calcium sensing receptor (CaSR), which in addition to parathyroids it is also contained in the basolateral side of the thick ascending loop of Henle cells. The hypercalcemia activate the CaSR of these cells which in turn inhibit the renal outer medullary potassium channel (ROMK) resulting in a decreases in the content of potassium to the tubular lumen and becomes less positively charged, thus calcium (positively charged) remains in the tubular lumen, it is not reabsorbed and circulates to the distal and collecting tubules to be excreted in urine.
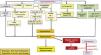
In addition, to prevent precipitation of this excess calcium in the tubule, the Na+/K+/2Cl- transporter (NKCC2) is also inhibited, which leads to an increase in sodium content in the tubular lumen, its increased excretion and arrival at the distal level, both to the intercalated cells and to the main cells of the cortical collecting tubule (CCT), which also have CaSR. At this level the activation of the CaSR stimulates proton transport in the intercalated cells, which acidifies the urine and also decreases aquaporins in the principal cells, resulting in more diluted urine. Furthermore, the non-reabsorption of sodium triggers, by hypovolemia-dependent mechanisms, the synthesis of aldosterone, which, through the mineralocorticoid receptor (MCR) in the principal and intercalated cells of the CT, stimulates, through phosphorylation of the Na+/K + ATPase pump, the apical sodium channels (ENAC), ROMK, H+-ATPase and H+/K+-ATPase. These changes lead to an increase in the excretion of protons into the lumen1 (Fig. 1). In short, in order to excrete excess calcium without crystal precipitation, a greater amount of urine is required, more diluted and more acidic, to which the calcium and sodium balance contributes and in which aldosterone, as the ultimate effector of the hormonal axis that includes renin and angiotensin II (ANG II), plays a major role in the balance of the effective circulating volume. Hence, in situations of hyperaldosteronism we will find volume-dependent hypertension, hypokalemia and metabolic alkalosis.

Classic relationship between calcium and sodium transport.
(1) Calcium is transported by the paracellular pathway in the thick ascending loop of Henle, and thus increases its concentration in the interstitium. (2) When its concentration rises, the calcium-sensitive receptor is stimulated, which inhibits the ROMK activity. (3) The lower potassium excretion establishes a lower electropositivity of the tubular fluid. (4) Lower electropositivity inhibits paracellular calcium transport. (5) Parallel to this, the high calcium concentration inhibits the NKCC2 transporter. (6) This action decreases sodium reabsorption. The overall effect is an increase in sodium and calcium at the tubular level reaching distal segments. (7) Calcium stimulates CaSr in the intercalated cells of the CCT. (8) The H+-ATPase pump increases proton excretion into the tubular lumen and (9) makes the urine more acidic, which hinders calcium precipitation. (10) It also stimulates CaSr in CCT principal cells, (11) which causes inhibition of aquaporins (AQP) and arginine vasopressin (AVP). (12) The overall effect is the achievement of more dilute urine. (13) The lower sodium reabsorption induces aldosterone stimulation by hypovolemia-dependent mechanisms, which sets in motion its classic action on ENaC and ROMK receptors. (14) Aldosterone is involved in stimulating AVP.
AQP: aquaporins; Ca+2: calcium; H+: protons; K+: potassium; Na+: sodium; MCr: mineralocorticoid receptor; CaSr: calcium-sensitive receptor.
Source: modified from Ranieri.1
PTH secretion is stimulated by hyperphosphatemia, hypocalcemia, the decrease in calcitriol and the increase in FGF23. HPT can cause a decrease in vitamin D receptors (VDR), resulting in a lower response to calcitriol and, secondarily, a greater secretion of PTH. PTH, through stimulation of the CyP27b1 gene, increases the synthesis of calcitriol and, therefore, the intestinal absorption of calcium, and at the bone, osteoclastogenesis, which causes the bone release of calcium and phosphorus. In the initial stages of chronic renal disease its overall effect is to increase phosphatemia, calcemia and phosphaturia and decrease calciuria, but as renal disease progresses and renal elimination of P decreases, phosphatemia will progressively increase, directly stimulating PTH secretion by post-transcriptional stimulation of the PTH gene, PT cell proliferation by activation of epidermal growth factor (EGFR) which causes TGF- αupregulation, inhibits CaSR expression and increases VDR expression in the intestine and decreases in the kidney.
Despite an extensive literature on the subject, the relationship between hyperaldosteronism (HA) and HPT is not completely clarified. Wermer2 and Sizemore et al.3, in 1963 and 1980, when defining MEN type 1 and 2 syndromes, already spoke of the relationship between the PT/adrenal axis. In the 1980s, the first cases of HA/HPT association were described,4–6 and since then the number of published cases has multiplied.7–11
What we should know about the close relationship between aldosterone and PTHALD and PTH levels are directly and bidirectionally related through mechanisms involving the ionic and acid/base balance of the internal environment.
ALD, through its actions on different tubular receptors, can induce hypercalciuria and chronically a negative calcium balance at a systemic level and hypocitraturia.
The first mechanism by which HA causes hypercalciuria depends on the expansion of circulating volume, which induces, through stimulation of tubulo-glomerular retrofiltration, a reduced reabsorption of sodium and calcium in the proximal tubule (PT), since both transports are coupled.12,13
However, in addition to chronic negative calcium balance, the mechanism by which HA can induce HPT appears to be mediated by other metabolic factors.
The expansion of circulating volume causes suppression of ANG II and increased expression of the ROMK channel, which determines hypokalemia due to increased renal excretion of potassium.
Hypokalemia favors intracellular acidosis, which, when becomes chronic, determines increased bicarbonate absorption at the proximal level and its reduced arrival at the distal level.
Therefore, in the HA there is a greater calcium supply and a lower bicarbonate supply at the distal level. Calcium should be reabsorbed through the apical calcium channel, but for it to function properly it needs to be stimulated by bicarbonate, and its lower arrival inactivates it, thus increasing hypercalciuria.
At the same time, acidosis in PT increases metabolism and citrate consumption, which will lead to hypocitraturia.
Complementarily, hypokalemia induces phosphate depletion in PT, with compensatory increase of calcitriol, which causes increased intestinal calcium absorption and decreased expression of aquaporins stimulating ammoniagenesis, which could result in chronic interstitial damage.
In addition, ALD stimulates the Na+/Cl- cotransporter in the distal tubule, which decreases calcium reabsorption in that segment and accentuates hypercalciuria and inhibition of the Na+/H + transporter, inducing incomplete distal renal tubular acidosis that boosts hypocitraturia.
Hypocitraturia, hypercalciuria and the negative calcium balance that would stimulate PTH secretion would increase the possibility of nephrolithiasis and medullary nephrocalcinosis14,15 (Fig. 2).

Generation of chronic renal damage.
Abs: absorption; ANG II: angiotensin II; AQP: aquaporins; TRA: tubulorenal acidosis; BIC: bicarbonate; Ca+2: calcium; H+: protons; Na+: sodium; P: phosphorus; MCr: mineralocorticoid receptor; DT: distal tubule; PT: proximal tubule; AT-1r: angiotensin I receptor; +: stimulus; >: major; <: minor.
It is hypothesized that an alteration of the calcium sensitive receptor (CaSR) would cause the calcium threshold for PTH synthesis by parathyroid cells to be higher than under normal conditions.16
A high percentage of studies show that calciuria decreases after treating HA, as we have seen in our case 1. The lack of unanimity is explained by the different stages of evolution of the disease in the reported cases.
It has been found that after treatment with R/ANG/ALD axis inhibitors in HA, PTH values decrease only when ALD levels are not elevated, which seems to demonstrate an effector mechanism of ALD in the control of PTH synthesis independent of renin and ANG II stimulation. ANG II increases PTH levels acutely through its direct action on the PT gland and chronically through the mineralocorticoid receptor (MCr) genomic pathway. With the chronic stimulus, secondary HPT becomes independent and can become tertiary, and latent HA can coexist with a clinically more evident hypercalcemia.17
Additionally, genes that reflect ALD synthesis are present in the metabolic pathways of bone metabolism and hypercalcemia activates CaSr.
PTH acts on ACTH receptors, on the R/ANG/ALD system and on calcium. The passage of calcium into the intracellular space exerts an initiating effect on the phosphorylation cascade that concludes in the activation of transcription factors (NURR1, NGF1B, CREB). These bind to the promoter region and positively regulate aldosterone secretion by stimulating the phospholipase C, protein kinase C and CYP11B2 transcription pathways (Fig. 3).

The action of PTH on the adrenal glands, calcium and R/ANG/ALD allows it to control ALD secretion at the adrenal glands. Both PTH and PTHr have secretagogue capacity in the adrenal gland.18,19
The fact that PTs possess adrenal metabolism receptors (MCr and ANGIIr) and adrenal PT receptors (PTH1 r, VDR and SCar), especially in cells of the zona glomerulosa,20,21 would speak in favor of a bidirectional regulatory pathway between bone metabolism and ALD (cytochrome p450 21 hydroxylase) adrenal steroidogenesis.
There are adjuvant factors that can influence in the behavior of the renal tubule in bone metabolism that are independent of this pathway, such as the salt-rich diet, which favors hypercalciuria by direct exchange and secondarily induces a stimulus for PTH secretion, independent of vitamin D levels; and vitamin D itself, which induces inhibition of ANG II and, by improving calcemia, has an inhibitory action on aldosterone through its effects on the vascular endothelium, but it has been shown in numerous studies that the HA/HPT ratio is independent of these factors.17
What we would like to knowAn interesting unanswered question is why calcium and sodium metabolism, carried out in the anatomically close Na+/Ca+2 and Na+/Cl- receptors in the distal tubule, and the synthesis of the hormones ALD and PTH are so intrinsically and undeniably linked. One possible explanation is circumstantial. Just as endocrine pathologies affecting different glands (PT, pituitary, pancreas, and adrenal) are found in MEN 1 and MEN 2 syndrome, there may be another undescribed multiple endocrine disorder that brings together PT and adrenal pathology in the clinical form described in these cases.
Another possible explanation would be related to an integrative conception of the balance of the internal environment, partially explained in the first section of this review. In the same way that it is essential to maintain stability between effective circulating volume and water balance, and this is achieved by means of a coordinated response of antidiuretic hormone and aldosterone,22 it is stimulating to think about the importance of extra- and intracellular calcium levels in the function regulation of the of blood vessels, and about the second messenger mechanisms that the ionic influx of calcium exerts directly in cells that may justify a common interrelation pathway between both systems, with which there would be inter-regulatory pathways between aldosterone and ADH balance and between aldosterone and PTH balance.
These two cases are illustrations of a specular reality, represent a primary HA due to bilateral adrenal hyperplasia that clinically presents with primary HPT that resolves when the HA is treated, and a primary HPT that, when treated, unmasks a subclinical HA that was not evident until that moment. The reality that this fact represents may have importance in the clinical practice of patients affected with both entities and should be taken into account when approaching the treatment of these patients.
Key concepts- -
ALD controls effective circulating volume and blood pressure. PTH controls bone mineral metabolism. Both hormones have common pathways of interrelation.
- -
HA induces hypercalciuria and hypocitraturia, which justifies a greater tendency to nephrolithiasis and medullary nephrocalcinosis by 5 interrelated mechanisms:
- 1
Lower distal tubular calcium absorption.
- 2
Hypokalemia, hypophosphatemia and increased synthesis of calcitriol.
- 3
Intracelular acidosis and hypocitraturia.
- 4
Incomplete distal tubulorenal acidosis.
- 5
ENaC receptor stimulation with volume expansion and angiotensin II suppression.
- -
HA causes chronic interstitial damage through ammoniogenesis-dependent mechanisms which involve the function of aquaporins.
- -
Hypercalciuria and chronic negative calcium balance in HA are direct stimuli for PTH secretion.
- -
PTH induces adrenal steroidogenesis by mechanisms dependent on calcium second messenger pathways, direct renin and ACTH stimulation.
This article has no sources of funding.
Conflicts of interestThe authors declare that they have no conflicts of interest.



