

Introducción. La hiperoxaluria primaria tipo 1 es una rara enfermedad de herencia autosómica recesiva, que está causada por mutaciones en el gen AGTX que codifica la enzima alanina glioxilato aminotransferasa que es necesaria en la metabolización del glioxilato. Una actividad insuficiente de esa enzima conduce a un incremento en la conversión de glioxilato a oxalato. Dado que el oxalato no se metaboliza en los humanos y se elimina por vía renal, el riñón es el primer órgano afectado, dando lugar a la aparición de litiasis de repetición, nefrocalcinosis e insuficiencia renal precoz.
Pacientes y métodos. Se estudia la evolución a largo plazo de 4 pacientes diagnosticados en la edad pediátrica de hiperoxaluria primaria tipo 1. Tres de ellos son adultos en la actualidad. Todos eran portadores en 1 o ambos alelos de la mutación I244T, que se asocia al polimorfismo P11L en el gen AGTX.
Resultados. La presentación clínica fue en la primera década de la vida, 3 con litiasis renal múltiple y 1 con nefrocalcinosis. Este último caso, que debutó en el período de lactante, ha manifestado un deterioro de la función renal más marcado en un tiempo más corto. Los otros 3 han mostrado una reducción lenta del filtrado glomerular y han formado escasos nuevos cálculos tras tratamiento con piridoxina, citrato potásico y sales de magnesio. Hasta el momento, no se han detectado signos de oxalosis sistémica.
Conclusiones. Los pacientes diagnosticados en la infancia que son portadores de la mutación “canaria” de hiperoxaluria primaria tipo 1 muestran una evolución aceptable en respuesta al tratamiento farmacológico, a excepción de que debuten con nefrocalcinosis.
INTRODUCCIÓN
La hiperoxaluria primaria (HP) se origina por un desorden metabólico hereditario autosómico recesivo del metabolismo del glioxalato, que causa una producción excesiva de oxalato. La HP tipo 1 (HP1) es una rara enfermedad autosómica recesiva, que se caracteriza por un defecto funcional de la enzima alanina glioxalato aminotransferasa (AGXT)1. Se expresa específicamente en el hígado y se localiza en el peroxisoma hepático, por lo que su disfunción resulta en una elevada producción hepática de oxalato2.
Dado que el oxalato no se metaboliza en los humanos y se elimina por vía renal, el riñón es el primer órgano afectado, dando lugar a la aparición de litiasis de repetición, nefrocalcinosis e insuficiencia renal precoz. Los pacientes, generalmente presentan los primeros síntomas de la segunda a la tercera década de la vida2. El cuadro clínico puede variar en gravedad de acuerdo con el tipo de mutación presente y a la presencia o no de ciertos polimorfismos asociados3.
En las Islas Canarias, la mutación más frecuente es la I244T que es patógena cuando está asociada al polimorfismo L11P. Fue descrita en un grupo de pacientes adultos en 2003 por Santana et al4. Hemos estudiado la evolución a largo plazo de 4 pacientes diagnosticados en la edad pediátrica portadores de la mutación “canaria” de HP1.
PACIENTES Y MÉTODOS
Se evaluaron las historias clínicas de 4 pacientes con diagnóstico de HP1 originarios de 2 de las Islas Canarias, a saber: La Palma (pacientes 3 y 4) y La Gomera (pacientes 1 y 2, hermanos). Estos están emparentados con el paciente 3, ya que la bisabuela paterna de los primeros era hermana del bisabuelo materno del segundo.
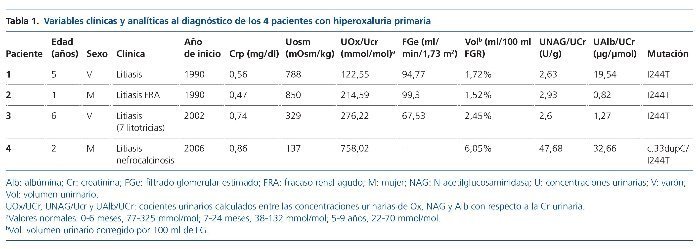
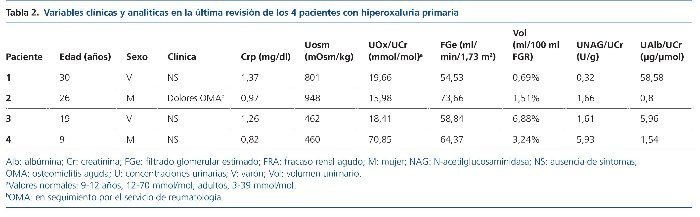
Hemos revisado la edad, el tipo de mutación presente en el gen AGTX, las manifestaciones clínicas y los datos analíticos y ecográficos al debut y al final del período de seguimiento. El seguimiento abarca desde el año 1990 (pacientes 1 y 2) hasta mayo de 2015 (tablas 1 y 2).
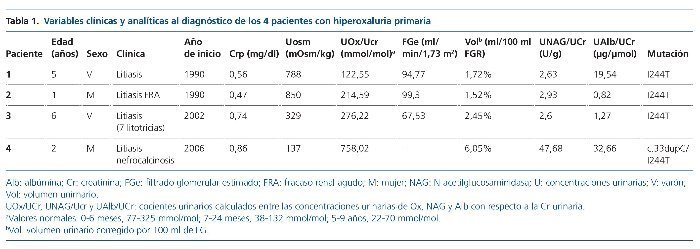
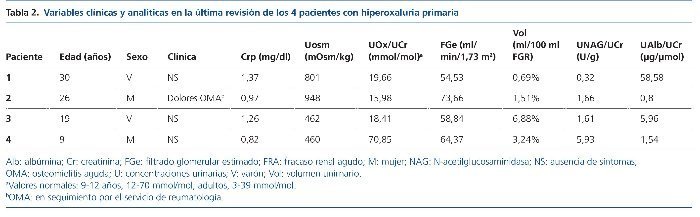
Se han recogido los datos de los valores del filtrado glomerular estimado (FGe), la osmolalidad urinaria máxima (Uosm) tras estímulo con desmopresina y los resultados de los cocientes urinarios, calculados en muestras de orina aislada, oxalato/creatinina, albúmina/creatinina y N-acetilglucosaminidasa/creatinina (NAG/creatinina).
El cálculo del FGe se realizó mediante la fórmula de Schwartz (ecuación modificada, 2009): 0,413 × talla (cm)/creatinina en plasma (Crp [mg/dl])5. Los pacientes se clasificaron en estadios según las guías KDIGO 20126. Se calculó, además, el volumen urinario corregido por 100 ml de FG según la fórmula: Crp × 100/creatinina en orina (Cru [mg/dl]).
RESULTADOS
Todos debutaron en la edad pediátrica. La edad promedio al momento del diagnóstico fue de 37,7 meses (2-76 meses) (tabla 1). El seguimiento se extendió hasta 25 años después del diagnóstico en los 2 primeros casos (pacientes 1 y 2). La presentación al debut estuvo relacionada con la presencia de litiasis sintomática (paciente 3), con un hallazgo ocasional (pacientes 1 y 2) o con rechazo de la alimentación e irritabilidad a los 51 días de vida (paciente 4); el hallazgo de leucocituria estéril motivó la solicitud de una ecografía renal, que mostró imágenes de nefrocalcinosis. La paciente 2, poco tiempo después de su diagnóstico, a los 22 meses de vida, sufrió un episodio de fracaso renal agudo anúrico obstructivo secundario a impactación litiásica en ambas uniones pieloureterales en el contexto de una infección por el virus de la varicela. Fue tratada mediante derivación urinaria con catéter doble J bilateralmente (fig. 1) y, después, sometida a litotricia.
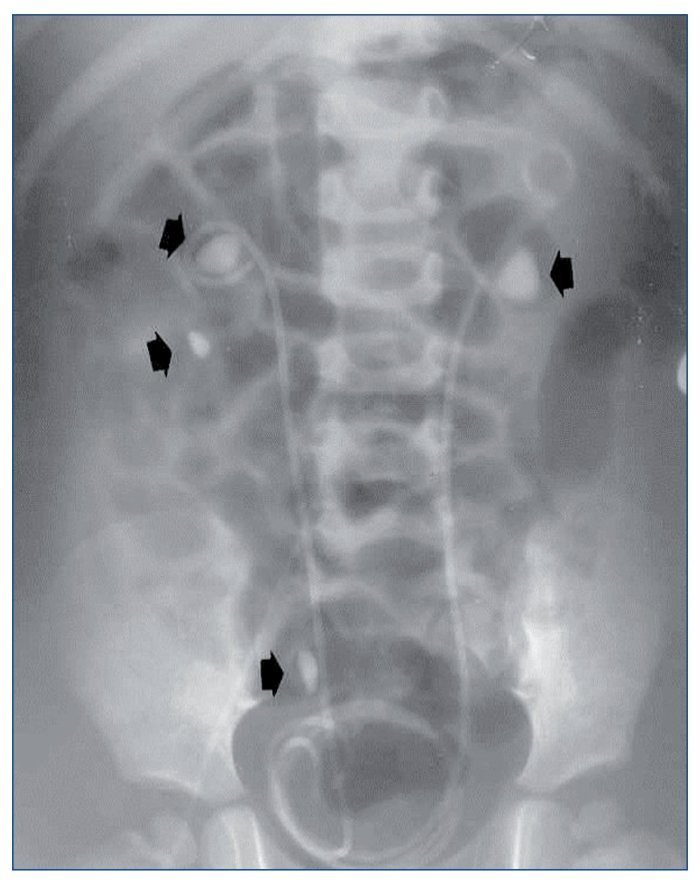
Figura 1. Obstrucción bilateral pieloureteral de origen litiásico (flechas), que produjo un cuadro de insuficiencia renal aguda a la paciente 2 cuando contaba 23 meses de edad. Precisó la colocación bilateral de catéter doble J. Obsérvense, además, sendos cálculos en el cáliz inferior y el uréter distal derechos (flechas).
En cuanto a la función renal al diagnóstico, además de la hiperoxaluria, los 4 pacientes tenían incremento del volumen urinario, 3 de ellos defecto de la capacidad de concentración y 2, elevación de los valores de creatinina (tabla 1). Los valores de los ácidos glicólico y L-glicérico fueron normales en los pacientes 1 y 2.
Al final del período de seguimiento, todos los pacientes mostraban enfermedad renal crónica (54,5-73,6 ml/min/1,73 m2). Tres mostraban incremento en el volumen urinario y 2 revelaban un defecto en la capacidad de concentración renal y albuminuria elevada. La eliminación urinaria de NAG estaba aumentada únicamente en la niña que debutó con nefrocalcinosis (tabla 2).
La excreción urinaria de oxalato está actualmente controlada en todos los casos con tratamiento médico a base de piridoxina. El tratamiento inhibidor de la cristalización se realizó con citrato potásico y sales de magnesio.
Tres de los pacientes son homocigotos para la mutación I244T acompañada del polimorfismo P11L. Solamente 1 paciente (caso 4), la de peor evolución, es heterocigota compuesta (c 33dupC/I244T) (tabla 2).
En ninguno de los pacientes se han detectado depósitos de oxalatos extrarrenales.
DISCUSIÓN
Se presenta el primer estudio evolutivo a largo plazo de 4 pacientes diagnosticados en edad pediátrica de HP1, portadores de la misma mutación procedentes de 3 familias canarias.
La prevalencia de HP1 estimada es variable, y oscila entre 1-2,9 pacientes por millón de habitantes. En las Islas Canarias se ha observado una elevada prevalencia de esta enfermedad. Se ha notado que un gran porcentaje de estos pacientes proceden de la isla de La Gomera7. Se ha comunicado que la consanguinidad, frecuente en esa isla en el pasado, está muy relacionada con la aparición de la enfermedad8. Es poco conocido que el primer niño diagnosticado de hiperoxaluria en España procedía de las Islas Canarias9.
Se han descrito más de 150 mutaciones distribuidas en los 11 exones del gen AGTX. La más frecuente a nivel mundial es la denominada p.Gly170Arg (G170R) (20-40% de los casos)3. No obstante, la inmensa mayoría de los pacientes canarios con HP1 posee la segunda mutación más frecuentemente comunicada (I244T), probablemente como consecuencia de un efecto fundador, ya que los haplotipos de las familias estudiadas comparten alelos de otros marcadores intragénicos4. La mutación consiste en un cambio T/C en el nucleótido 853 (exon 7), que corresponde a un cambio Ile por Thr en el codón 244 de la proteína AGTX (I244T). Aunque el cambio I244T, aisladamente, no afecta la actividad AGTX o su localización subcelular, cuando está presente en la misma molécula el polimorfismo común Pro-11 a Leu-11 (P11L) se produce una modificación intragénica que da lugar a una proteína mal plegada, que tiende a formar agregados inactivos con una menor actividad enzimática en los extractos celulares solubles4.
La mutación I244T en combinación con el polimorfismo P11L en el gen AGTX demuestra que la HP1 es una enfermedad conformacional que podría ser susceptible de tratamiento con chaperonas farmacológicas4. Por el contrario, la mutación c.33dupC, presente en un alelo de la paciente 4, conlleva un defecto absoluto de síntesis, que se refleja en un fenotipo más agresivo en este caso, con nefrocalcinosis infantil.
La totalidad de nuestros casos han desarrollado un deterioro lento pero progresivo de la función renal y 2 de ellos están encuadrados en el estadio 2 de enfermedad renal crónica (casos 2 y 4) y otros 2, en el estadio 3 (casos 1 y 3) (tabla 2).
El tratamiento de la HP1 debe ser precoz y dirigirse a disminuir la saturación urinaria de oxalato cálcico, aumentando la ingesta de líquidos y empleando inhibidores urinarios de la cristalización. La piridoxina (vitamina B6) es, tal vez, la única medida capaz de reducir de forma eficaz la producción de oxalato, aunque solo es aplicable en los casos de HP11. En nuestros casos, es llamativa la buena respuesta al tratamiento (tabla 2).
Una vez que el diagnóstico de HP1 es firme, el tratamiento prioritario y potencialmente curativo es el trasplante hepático. Dado que lo habitual es estar ante un deterioro renal avanzado e irreversible, la indicación es el trasplante doble, hepático y renal, el cual se debe programar precozmente, con FGe entre 15 y 30 ml/min/1,73 m2, para prevenir el depósito tisular de oxalato11.
Salido et al han generado un modelo murino de hiperoxaluria (Agtx KO), así como ratones transgénicos que expresan los haplotipos con las mutaciones más frecuentes de la HP1. En estos modelos animales han evaluado estrategias terapéuticas como las terapias enzimática, celular y génica. Entre los vectores de terapia génica probados, los virus adeno asociados tipo 8 (AAV8) que expresan el cADN del gen AGTX humano han sido capaces de corregir el defecto enzimático en los ratones Agtx KO, proporcionando una reserva funcional similar o incluso superior a la de los animales control, sin un defecto enzimático12,13.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Correspondencia:
José Carlos Rodríguez-Pérez
Servicio de Nefrología. Hospital Universitario de Gran Canaria Dr. Negrín.
35019 Las Palmas de Gran Canaria.
jrodperd@gobiernodecanarias.org


