
Hereditary renal hypouricemia is a rare autosomal recessive genetic disorder that involves an isolated defect in uric acid reabsorption at the renal tubules. Patients present with serum uric acid concentrations of less than 2mg/dl (119μmol/L) with increased fractional excretion above 10%. Most of the patients are asymptomatic and are detected incidentally. However, complications such us nephrolithiasis, hematuria, acute renal failure exercise-induced or after dehydration for acute gastroenteritis, or posterior reversible encephalopaty syndrome (PRES) may develop.
Hereditary renal hypouricemia is confirmed by molecular genetic analysis of the two genes which codify the uric acid transport in the kidney tubules. The renal hypouricemia type 1 (OMIM 220150) is characterised by loss-of-function mutations in the SLC22A12 gene which encodes URAT 1 transporter, and the hypouricemia type 2 (OMIM 612076) is caused by defects in the SLC2A9 gene. Homozygous mutations of SLC2A9 cause the most severe forms of the disease.
Most mutations have been identified in Japanese adults, and only a few in children.
We describe three asyntomatic paediatric Spanish patients with renal hypouricemia, with genetic confirmation, and we make a revision of all of the paediatric cases with genetic study published in the literature.
La hipouricemia renal hereditaria es un trastorno genético, poco frecuente, causado por un defecto aislado en la reabsorción del ácido úrico a nivel del túbulo renal. Los pacientes presentan concentraciones séricas de ácido úrico inferiores a 2mg/dl (119 micromol/L), y un incremento en la excreción fraccional de ácido úrico mayor del 10%. La mayoría son asintomáticos y se detectan accidentalmente, aunque pueden aparecer complicaciones como la nefrolitiasis, hematuria, daño renal agudo inducido por ejercicio físico o tras un episodio de deshidratación por gastroenteritis aguda, o el síndrome de encefalopatía posterior reversible.
La hipouricemia renal hereditaria se confirma por el análisis molecular de los dos genes que codifican los transportadores de urato a nivel del túbulo renal. La hipouricemia renal tipo 1 (OMIM 220150) con pérdida de función en el gen SLC22A2 que codifica el transportador URAT1 y la hipouricemia renal tipo 2 (OMIM 612076) con mutaciones en el gen SLC2A9 que codifica el transportador GLUT9. Las formas más graves se producen en pacientes con mutaciones en el gen SLC2A9 en homocigosis. La mayoría de mutaciones se han descrito en adultos Japoneses, y sólo unos pocos casos en niños. Presentamos tres casos de niños españoles asintomáticos con hipouricemia renal confirmada genéticamente y realizamos revisión de los casos pediátricos con estudio genético, publicados en la literatura.
Hereditary renal hypouricaemia (HRH) is a rare and underdiagnosed genetic disorder, and is included in the group of rare diseases (ORPHA 94088). It is caused by an isolated defect in the renal transport of uric acid at the renal tubular level, either due to decreased reabsorption and/or increased secretion.1,2 In children over 1 year old and in adults, it must be suspected in the presence of persistent serum uric acid (UA) level of less than 2mg/dl (119μmol/l), with fractional excretion of UA (FEUA) above 10% (normal 7.25±2.98%).3–5 HRH is confirmed by the molecular analysis of the 2 known genes that encode UA transporters at the tubular level: SCL22A12 and SLC2A9.4,6SLC22A12 encodes the URAT1 transporter located in the apical membrane of the proximal tubule (PT). mutations of this transporter, with an autosomal recessive inheritance pattern, are responsible for HRH type 1 (OMIM 220150).7SLC2A9 encodes 2 isoforms of the GLUT9 transporter, 1 long and 1 short, located respectively in the basolateral membrane of the PT and in the apical membrane of the collecting tubule; mutations, with an autosomal recessive, and some cases dominant, inheritance pattern, are responsible for HRH type 2 (OMIM 612076).8,9
Most patients are asymptomatic, especially during childhood, so many cases are not diagnosed or are discovered accidentally. Occasionally, the first manifestation may be haematuria or hypercalciuria, but diagnosis is usually made when HRH-related complications appear, such as nephrolithiasis, acute kidney injury (AKI) induced by intense or less frequent physical exercise, after an episode of dehydration due to acute rotavirus gastroenteritis10–17 or posterior reversible encephalopathy syndrome (PRES) in patients with exercise-associated AKI.18–20
The prevalence of HRH is unknown, and there are only some data on prevalence of hypouricaemia.21–24
More than 150 patients with HRH type 1 have been reported in the literature, mostly Asian adults (Japan, South Korea), with the most frequent mutation (2.30–2.37%) being p.W258X. Only a small number of cases with HRH type 2 have been published.25–30
Recent studies show that this disease is not limited to the Asian population, since genetic studies have made diagnosis possible in European patients from different ethnic groups.6,27–29,31
Forty-seven mutations of the SCL22A12 gene have been described worldwide (36 of them reversing or non-reversing) and 25 of the SLC2A9 gene (15 of them reversing or non-reversing), of which 30 and 13 produce disease, respectively.32
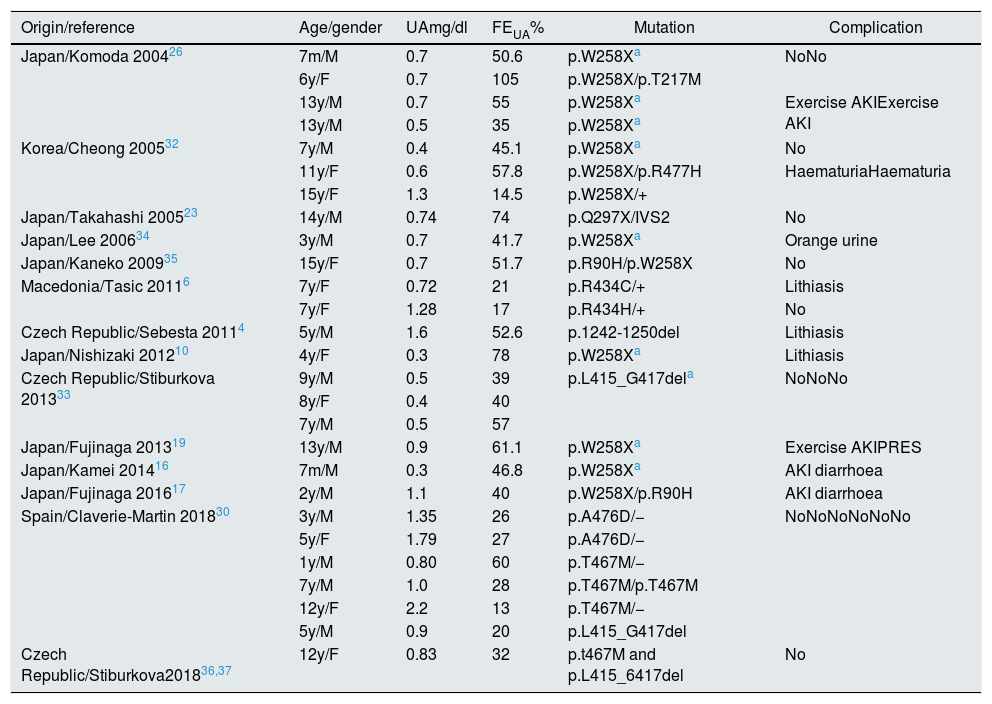
We performed a literature review of paediatric cases published with genetic study. The analytical data, the mutations found, and the most relevant clinical manifestations are shown in Tables 1 and 2.4,6,10,12,13,16–20,23,26,27,30,32–39 In total, 27 children with HRH type 1 and 14 with HRH type 2 were identified, of which 9 cases are Spanish.30
Children with HRH type 1: mutations described in SLC22A12.
| Origin/reference | Age/gender | UAmg/dl | FEUA% | Mutation | Complication |
|---|---|---|---|---|---|
| Japan/Komoda 200426 | 7m/M | 0.7 | 50.6 | p.W258Xa | NoNo |
| 6y/F | 0.7 | 105 | p.W258X/p.T217M | ||
| 13y/M | 0.7 | 55 | p.W258Xa | Exercise AKIExercise AKI | |
| 13y/M | 0.5 | 35 | p.W258Xa | ||
| Korea/Cheong 200532 | 7y/M | 0.4 | 45.1 | p.W258Xa | No |
| 11y/F | 0.6 | 57.8 | p.W258X/p.R477H | HaematuriaHaematuria | |
| 15y/F | 1.3 | 14.5 | p.W258X/+ | ||
| Japan/Takahashi 200523 | 14y/M | 0.74 | 74 | p.Q297X/IVS2 | No |
| Japan/Lee 200634 | 3y/M | 0.7 | 41.7 | p.W258Xa | Orange urine |
| Japan/Kaneko 200935 | 15y/F | 0.7 | 51.7 | p.R90H/p.W258X | No |
| Macedonia/Tasic 20116 | 7y/F | 0.72 | 21 | p.R434C/+ | Lithiasis |
| 7y/F | 1.28 | 17 | p.R434H/+ | No | |
| Czech Republic/Sebesta 20114 | 5y/M | 1.6 | 52.6 | p.1242-1250del | Lithiasis |
| Japan/Nishizaki 201210 | 4y/F | 0.3 | 78 | p.W258Xa | Lithiasis |
| Czech Republic/Stiburkova 201333 | 9y/M | 0.5 | 39 | p.L415_G417dela | NoNoNo |
| 8y/F | 0.4 | 40 | |||
| 7y/M | 0.5 | 57 | |||
| Japan/Fujinaga 201319 | 13y/M | 0.9 | 61.1 | p.W258Xa | Exercise AKIPRES |
| Japan/Kamei 201416 | 7m/M | 0.3 | 46.8 | p.W258Xa | AKI diarrhoea |
| Japan/Fujinaga 201617 | 2y/M | 1.1 | 40 | p.W258X/p.R90H | AKI diarrhoea |
| Spain/Claverie-Martin 201830 | 3y/M | 1.35 | 26 | p.A476D/− | NoNoNoNoNoNo |
| 5y/F | 1.79 | 27 | p.A476D/− | ||
| 1y/M | 0.80 | 60 | p.T467M/− | ||
| 7y/M | 1.0 | 28 | p.T467M/p.T467M | ||
| 12y/F | 2.2 | 13 | p.T467M/− | ||
| 5y/M | 0.9 | 20 | p.L415_G417del | ||
| Czech Republic/Stiburkova201836,37 | 12y/F | 0.83 | 32 | p.t467M and p.L415_6417del | No |
AKI: acute kidney injury; F: female; FEUA: fractional excretion of uric acid; M: male; m: months; PRES: posterior reversible encephalopathy syndrome; UA: serum uric acid; y: year.
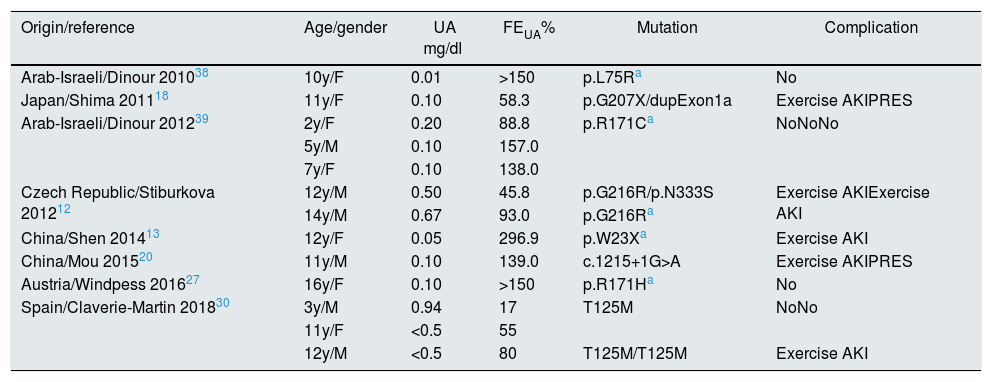
Children with HRH type 2: mutations described in the SLC2A9 gene.
| Origin/reference | Age/gender | UA mg/dl | FEUA% | Mutation | Complication |
|---|---|---|---|---|---|
| Arab-Israeli/Dinour 201038 | 10y/F | 0.01 | >150 | p.L75Ra | No |
| Japan/Shima 201118 | 11y/F | 0.10 | 58.3 | p.G207X/dupExon1a | Exercise AKIPRES |
| Arab-Israeli/Dinour 201239 | 2y/F | 0.20 | 88.8 | p.R171Ca | NoNoNo |
| 5y/M | 0.10 | 157.0 | |||
| 7y/F | 0.10 | 138.0 | |||
| Czech Republic/Stiburkova 201212 | 12y/M | 0.50 | 45.8 | p.G216R/p.N333S | Exercise AKIExercise AKI |
| 14y/M | 0.67 | 93.0 | p.G216Ra | ||
| China/Shen 201413 | 12y/F | 0.05 | 296.9 | p.W23Xa | Exercise AKI |
| China/Mou 201520 | 11y/M | 0.10 | 139.0 | c.1215+1G>A | Exercise AKIPRES |
| Austria/Windpess 201627 | 16y/F | 0.10 | >150 | p.R171Ha | No |
| Spain/Claverie-Martin 201830 | 3y/M | 0.94 | 17 | T125M | NoNo |
| 11y/F | <0.5 | 55 | |||
| 12y/M | <0.5 | 80 | T125M/T125M | Exercise AKI |
AKI: acute kidney injury; F: female; FEUA: fractional excretion of uric acid; M: male; m: months; PRES: posterior reversible encephalopathy syndrome; UA: serum uric acid; y: year.
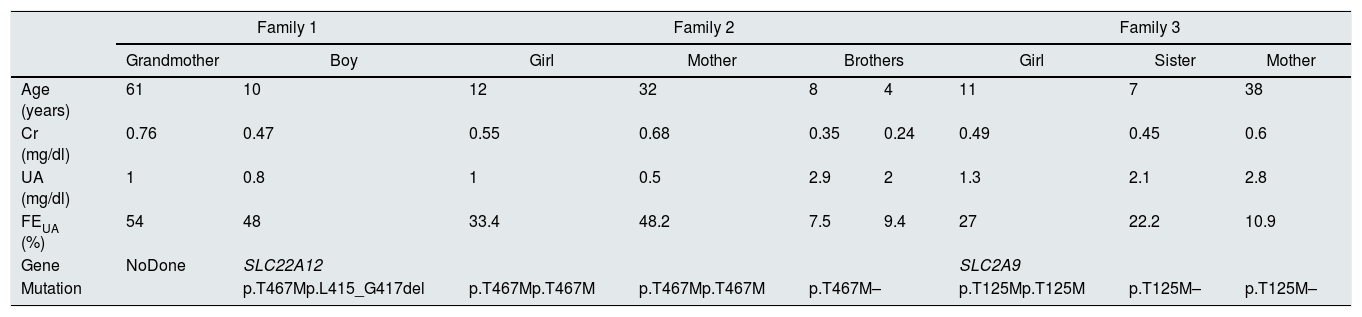
We present the cases of 2 children with HRH type 1, from 2 unrelated Roma families, and 1 Caucasian child with HRH type 2 (Table 3).
Clinical and genetic data of the patients and their families.
| Family 1 | Family 2 | Family 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Grandmother | Boy | Girl | Mother | Brothers | Girl | Sister | Mother | ||
| Age (years) | 61 | 10 | 12 | 32 | 8 | 4 | 11 | 7 | 38 |
| Cr (mg/dl) | 0.76 | 0.47 | 0.55 | 0.68 | 0.35 | 0.24 | 0.49 | 0.45 | 0.6 |
| UA (mg/dl) | 1 | 0.8 | 1 | 0.5 | 2.9 | 2 | 1.3 | 2.1 | 2.8 |
| FEUA (%) | 54 | 48 | 33.4 | 48.2 | 7.5 | 9.4 | 27 | 22.2 | 10.9 |
| Gene | NoDone | SLC22A12 | SLC2A9 | ||||||
| Mutation | p.T467Mp.L415_G417del | p.T467Mp.T467M | p.T467Mp.T467M | p.T467M– | p.T125Mp.T125M | p.T125M– | p.T125M– | ||
Cr: plasma creatinine; FEUA: fractional excretion of uric acid; UA: serum uric acid.
Family 1. A 10-year-old child with no personal history of interest; weight and height in p50. Non-consanguineous parents, of romanian ethnicity, with no known history nephrourological diseases. Brothers 8 and 4 years of age, healthy. Paternal grandparents, first cousins. The paternal grandfather reported renal stones. In a control analysis of treatment with methylphenidate, a serum UA of 0.6mg/dl was observed on 2 occasions. In a lab work conducted 4 years a go due to anorexia, the value of UA was 0.7mg/dl, which was “unnoticed”. After the diagnosis of sustained hypouricaemia in an asymptomatic child with normal transaminases, renal function was studied. Glycosuria, proteinuria, and hypercalciuria were not detected in an isolated sample of urine, and the FEUA was 48%. The acid-base balance and renal ultrasound were normal. A review of the family clinical history showed that the paternal grandmother had a sustained hypouricaemia of 0.9–1.1mg/dl, which was not evaluayed, and the FEUA was 54%. Given the suspicion of HRH, and having obtained informed consent, a genetic study was carried out in the child, finding 2 heterozygosis mutations in SLC22A12 (p.T467M and p.L415_G417del). The grandmother did agree to have a genetic study, and it was not possible to study the parents and siblings either, because they lived in another community.
Family 2. A 12-year-old child with no personal history of interest; weight in p97 and height in p50. Maternal grandparents and parents, first cousins, of romanian ethnicity. Mother, grandmother and maternal aunt with history of renal stones. Brothers without heatlh problems. At 3 years of age, during a study for anorexia, iron deficiency anaemia and a serum UA of 0.4mg/dl were detected. Six months after treatment with oral iron, a control analysis showed persistent hypouricaemia (0.5mg/dl). A urine sample did not show glucosuria, proteinuria or hypercalciuria, and the FEUA was 12%. The patient moved to another region and no further studies were performe. At the age of 10 y.o. she came back asymptomatic, with serum UA concentration of 1mg/dL and FEUA of 33%. The diagnostic tests were completed in the patient and mother who, according to her history, also presented persistent hypouricaemia, serum UA of 0.5–0.6mg/dl, not evaluated, despite renal stones. The acid-base balance and renal ultrasound were normal for both. With the suspicion of HRH, and having obtained informed consent, a study was also requested of the brothers, who had normal serum UA levels. The genetic study showed that the mother and daughter were homozygous and the 2 brothers are heterozygous for the p.T467M mutation of the SLC22A12 gene. The maternal aunt, who had hypouricaemia of 0.6mg/dl, and the grandmother did not consent genetic analysis.
Family 3. An 11-year-old girl with a history of episode of pyelonephritis; weight and height in p50. Non-consanguineous parents, of Caucasian ethnicity, with no known nephrourological disease. Sisters 7 and 2 years of age, healthy. In the blood test performed during the episode of pyelonephritis and during its follow-up, serum UA levels were between 1.2 and 1.8mg/dL. These data initially went “unnoticed”. A urine sample did not show glucosuria, proteinuria or hypercalciuria, and the FEUA was 27%. Given the suspicion of HRH due to asymptomatic hypouricaemia with increased FEUA, after informed consent was obtained, a genetic study was performed, which demonstrated a homozygosis mutation of the SLC2A9 gene (p.T125M). The study was extended to first-degree relatives; the mother and the 7-year-old sister are carriers of the homozygosis mutation.
Mutational analysis of genes SLC22A12 and SLC2A9Using peripheral blood samples from the patients and their relatives, genomic DNA was isolated using a commercial kit (Gen Elute Blood Genomic DNA kit, Sigma-Aldrich, St. Louis, MO, USA). The coding exons of SLC22A12 and SLC2A9 were amplified by PCR and analysed by automatic sequencing with the BigDye Terminator v3.1 Cycle Sequencing kit in the 3500 Series Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The DNA sequences were compared with their respective reference sequences (SLC22A12, NCBI: NG_008110.1; SLC2A9, NCBI: NG_011540.1).
CommentsThis is the only study that includes a bibliographic review of all paediatric cases of HRH, and the first study in Spain of genetically confirmed paediatric cases. We present two romaniar children with HRH type 1: one compound heterozygous child (p.T467M/p.L415_G417) and one homozygote child (p.T467M/p.T467M), along with 2 brothers who were heterozygous for the p.T467M/+ mutation. Both mutations have been described previously in children and adults of Roma ethnicity in Spain and the Czech Republic.28–30 The frequency of these mutations in the Spanish romanian cohort is greater than in the Czech Republic (9.19% and 4.17% vs. 5.56% and 1.87%), and are the highest in the world, suggesting a high incidence of HRH in this ethnic group with frequent inbreeding and high incidence of kidney stones.29 Recently, 3 Spanish children with the p.T467M mutation have been described, 1 homozygote and 2 heterozygotes, together with 3 adult homozygous siblings for this mutation.30
The girl with HRH type 2, of Caucasian race, presents the p.T125M mutation in homozygosis in the SLC2A9 gene, while the sister and the mother are carriers in heterozygosis. This mutation was first identified in an 84-year-old Sephardic-Jewish adult, and later in 3 Spanish children, 2 of them asymptomatic and 1 who presented AKI induced by intense physical exercise.20,30
Knowledge of the ethnic group is important in the diagnosis and to facilitate the search for the type of mutation.
In children, hypouricaemia should be defined as a serum UA concentration of less than 2mg/dl in patients more than 1 year old. The fact is that between 2 and 12 months of age, due to “tubular immaturity”, FEUA is higher (between 27±21%)4 and the serum UA level is relatively lower (between 2.2 and 2.5mg/dl). After 1 year of age, the FEUA decreases to 8±6% and the serum UA increases to between 3.5 and 4.5mg/dl. These levels are maintained until 12 years of age, and from that point, levels are similar to those of an adult.4,5
We would like to draw attention to the importance of hypouricaemia, since due the fact that it does not have recognisable symptoms, often go unnoticed or are attributed to a laboratory error. All low levels of UA should be evaluated. The differential diagnosis is made on the basis of FEUA3; if it is increased (greater than 10%), it is of renal tubular origin, either in the form of a complex tubulopathy (primary or secondary Fanconi syndrome) or an isolated tubulopathy (HRH).
Although all our patients are asymptomatic, and were diagnosed fortuitously, HRH is known to present considerable clinical variability. Only 10% of patients with mutations in theSLC22A12 gene present clinical symptoms, while patients with homozygosis mutations in the SLC2A9 gene may present more severe symptoms due to a higher FEUA (even greater than 150%).8,9
Nephrolithiasis occurs in 10% of adults with a defect in the URAT1 transporter, and in 40% of those with an alteration in the GLUT9 transporter.3 This complication has been reported in 3 paediatric patients with mutation in the SLC22A12 gene.4,6,10
The sudden onset of AKI a few hours or days after performing physical exercise, such as a short distance run, or after an episode of acute rotavirus gastroenteritis, should guide the diagnosis of HRH. This complication has been reported in 4 children with a mutation in the SLC22A12 gene16,17,19,26 and 5 in the SLC2A9 gene, 1 of them being a 12-year old Spanish Caucasian.12,13,18,20,30 The initial symptoms are usually lumbar or abdominal pain together with nausea and muscle fatigue, which can be confused with a viral infection causing a delay of the diagnosis. The possibility of this disease should be considered in patients with acute renal failure and a “relatively low” serum UA concentration. Previous determinations of UA should be investigated and these should be repeated after renal function is normal, which in most cases occurs after a few days without dialysis.3
The proposed pathogenic mechanisms of AKI associated with exercise in HRH are:
- 1.
Increased production of UA during exercise which precipitates elimination in a more concentrated urine (due to hypovolaemia related to low fluid intake) which is also acidic (due to exercise) which facilitates UA precipitation with intratubular obstruction.40
- 2.
Reduced antioxidant capacity, due to hypouricaemia, allows the increase of free radicals during physical exercise to cause endothelial dysfunction and vasoconstriction of the renal arteries.41
- 3.
In patients with loss of urate transporter function (URAT 1 and GLUT 9), both tubular reabsorption of UA and secretion of organic anions to the tubular lumen are reduced resulting in PT cells damage.38
The importance of diagnosing this complication is to establish strategic measures to prevent recurrences: limit anaerobic physical exercise, maintain adequate hydration (before, during and after physical exercise) with abundant water intake, administration of antioxidants (vitamin C, carotenes), and even treatment with allopurinol.37
Two mechanisms by which allopurinol would act have been described:
- 1.
Decrease in UA production, with a lower amount of filtered UA, decreasing the risk of intratubular precipitation of UA.40
- 2.
Improvement of endothelial damage by reducing vascular oxidative stress.42
Posterior reversible encephalopathy syndrome (PRES) is a clinical-radiological entity characterised by headache, decreased level of consciousness, seizures and visual disturbances, with cerebral magnetic resonance images of cerebral oedema, more intense in the parieto-occipital white matter. It has been described in patients with HRH and AKI associated with exercise, and recently in paediatric patients: a 13-year-old boy with a mutation in the SLC22A12 gene and two 11-year-olds with a mutation in the SLC2A9 gene.18–20 It is thought that the presence of a sudden increase in blood pressure could be responsible for cerebral vasogenic oedema. Early diagnosis, along with adequate control of seizures and hypertension, avoiding an excess of intravenous fluids, are fundamental to prevent the appearance of neurological sequelae.43
Key concepts- 1.
Hypouricaemia is a finding d that often goes unnoticed or is attributed to a laboratory error. All hypouricaemia below 2mg/dl should be investigated.
- 2.
In children over 1 year old with UA<2mg/dl and FEUA>10%, HRH should be suspected.
- 3.
Diagnosis of HRH is confirmed through the molecular testing of 2 genes, SLC22A12 (HRH type 1) and SLC2A9 (HRH type 2).
- 4.
Most patients with HRH are asymptomatic. The most common complications are macro- or microscopic haematuria, hypercalciuria, lithiasis and AKI after physical exercise.
- 5.
To avoid complications and the recurrence of AKI, it is recommended abundant fluids intake before, during and after exercise, as well as treatment with allopurinol.
The authors have no conflicts of interest to declare.
The genetic analysis was funded by PI14/00760, part of the 2013–2016 National R&D&I Plan and co-funded by the ISCIII-Subdirección General de Evaluación y Fomento de la Investigación [General Subdirectorate for Evaluation and Promotion of Research] and the European Fund for Regional Development “A way to make Europe”.
Please cite this article as: Peris Vidal A, Marin Serra J, Lucas Sáez E, Ferrando Monleón S, Claverie-Martin F, Perdomo Ramírez A, et al. Hipouricemia renal hereditaria tipo 1 y 2 en tres niños españoles. Revisión de casos pediátricos publicados. Nefrologia. 2019;39:355–361.



