


Fibronectin glomerulopathy (FNG) is an uncommon autosomal dominant disease with age-related penetrance. Burgin1 first reported the diease in 1980. Strøm2 verified fibronectin glomerulopathy – a newly recognized autosomal dominant kidney disease. It has been distinguished from these other disease entities by the presence of glomerular fibrillary deposits that show strong immune reactivity to fibronectin (Fn).
Here we report a case of FNG 2017 in our hospital. A 45-year-old Chinese female was hospitalized because of edema for 7 days. She denied history of hair loss, oral ulceration, alopecia, oliguria, joint pain, rashes and menstrual irregularities. She had medical history of malignant hydatidiform mole 8-years ago which had been cured. Her sister had nephritic syndrome for ten years with no biopsy performed. Physical examination showed mild hypertension (158/119mmHg) and leg edema. The laboratory examination showed her daily urinary protein, serum albumin, blood urea nitrogen and creatinine were 4.21g/day, 40.9g/L, 4.39mmol/L, 53μmol/L. The blood tests of liver function, lipid, immunoglobulin, complement, antinuclear antibody spectrum, antineutrophil cytoplasmic antibody, hepatitis B and C serology were all negative. The renal and gynecologic ultrasound was normal. The kidney biopsy was showed in Fig. 1. Light microscopy showed one of 25 glomeruli was sclerotic glomeruli, others showed moderate to severe mesangial hypercellularity with a lobular appearance with cellular mesangial nodules that were expanded by matrix. Capillary walls showed stenosis, occlusion and basement membrane thickening. Segmental mesangial insertion and double track formation could be seen. Immunofliorescence only showed faint staining for IgM deposits in a band pattern in the subendothelial spaces and testing of IgG, IgA, C3, Cq1, fibrinogen, albumin completely negative. There was no evidence of amyloid deposits on Congo Red stain. Diffuse strong granular-smudgy staining was observed in mesangial and subendothelial locations with fibronectin. Electron microscopy showed 2 glomeruli that the capillary loops were lobulated, and focal endothelial cells proliferated. The wall of renal capsule is thickened and stratified. Extensive fine granular deposits in the mesangium and in the subendothelial spaces could be seen with a diameter of 10–12nm fibrils.
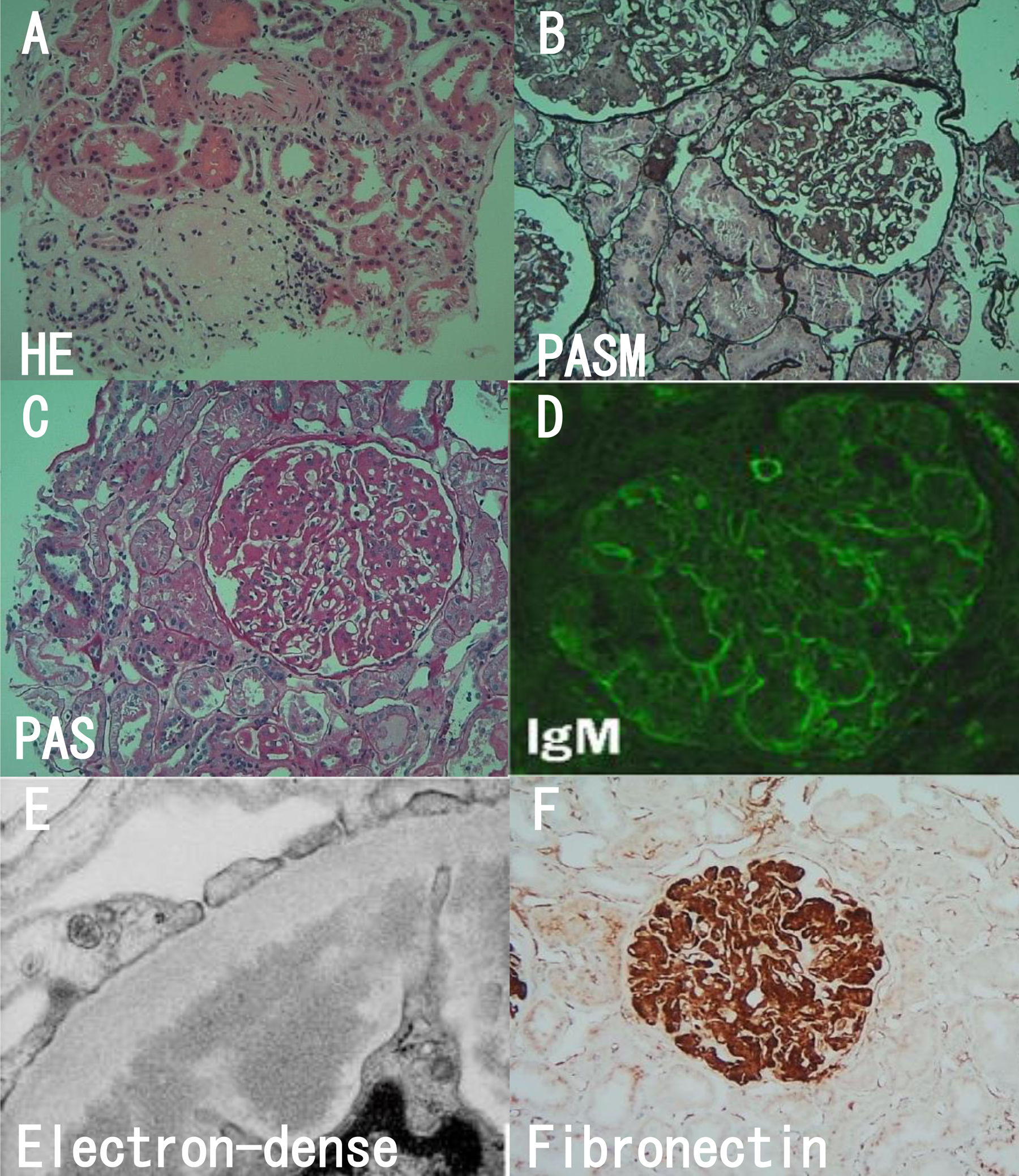
Light microscopic, immunofluorescence and electron studies. (A) Sclerotic glomeruli (HE). (B) Lobular appearance with cellular mesangial nodules (PASM). (C) Moderate to severe mesangial hypercellularity (PAS). (D) Faint staining for IgM (Immunofluorescence). (E) Massive electron-dense deposits. (F) Fibronectin was strongly positive (immunohistochemistry).
A PubMed and CNKI searching using the terms “fibronection glomerulopathy” yielded 35 English and 6 Chinese articles reports of patients with clinical and (or) biopsy characteristics until January 31, 2018. This allowed us to compare the disease clinical features and biopsy features. A total of 86 patients, 52 male, 34 female, aged 3–883 years were reported in these literatures. 25% of patients receive renal replacement therapy.2 42 patients among 78 patients observed with nephritic range proteinuria (more than 3.5g/day). 53 patients among 85 patients suffered from hypertension. 23 of 43 cases which were available Urine Analysis had microhematruia. 23 cases’ serum creatinine (normal values 0.6–1.3mg/dl) was abnormal except for 18 patients who did not provide renal function. Family history was noted in 61 patients. 57 patients of the literatures were provided with renal biopsy. 47 patients were tested intense staining for the serum fibronectin by immunohisto-chemistry which was the basis for the diagnosis. The Congo red staining is negative.
FNG is a newly recognized autosomal dominant hereditary renal disease. The deposition of Fn in the glomeruli is the key to FNG. The increasing Fn leads to some glomerular diseases. FNG is considered to be a hereditary disease and associated with mutations in the Fn1 gene.4,5 By haplotype analysis, p.Pro1472del and p.Leu1974Pro are fundamental mutations.6 FNG is considered an unimmune glomerulopathy. Diagnosis of FNG is made only by renal biopsy. Light microscopy has no diagnostic value for FNG. The diameter of the special deposits is important to diagnose in electron microscope. Fn staining of immunofluorescence can make a definite diagnosis. The disease is characterized by proteinuria, microscopic hematuria, hypertension and slow progression to end-stage renal failure. Some treatments such as glucocorticoid,7 immunosuppressant,8 ACEI, ARB9 et al. have used for the disease, but none of them is effective. There is still a risk of recurrence after renal transplantation.10
Our patient had the distinguishing feature of the disease on the basis of clinical symptoms: hypertension, proteinuria, microhematruia, family history and pathological manifestation: intense staining for the serum fibronectin, Congo red staining negative. At present, there are no specific treatments for FNG. Our patient had received angiotensin receptor blocker (ARB) and Shenyanshu tablets. After 3 months of follow-up, the urine protein was 2.85g/day. The proteinuria became less. However, further observation was needed to the proteinuria and renal function.
Conflict of interestThe authors declare that they have no conflict of interest.



