



This study screened for Fabry disease (FD) in patients in hemodialysis (HD) in the region of Madrid (CAM) with a cross-sectional design to evaluate HD-prevalent patients, followed by a three-year period prospective design to analyze HD-incident patients.
Inclusion criteriapatients older than 18 years on HD in the CAM, excluding patients diagnosed with any other hereditary disease with renal involvement different from FD, that sign the Informed Consent (IC). Exclusion criteria: underaged patients or not agreeing or not being capable of signing the IC.
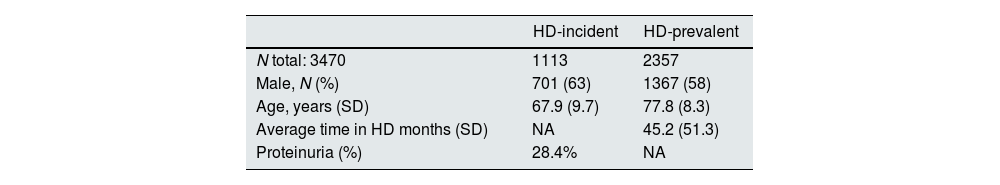
Results3470 patients were included, 63% males and with an average age of 67.9±9.7 years. 2357 were HD-prevalent patients and 1113 HD-incident patients. For HD-prevalent patients, average time in HD was 45.2 months (SD 51.3), in HD-incident patients proteinuria was present in 28.4%. There were no statistical differences in plasmatic alpha-galactosidase A (α-GAL-A) activity or Lyso-GL-3 values when comparing HD-prevalent and HD-incident populations and neither between males and females. A genetic study was performed in 87 patients (2.5% of patients): 60 male patients with decreased enzymatic activity and 27 female patients either with a decreased GLA activity, increased Lyso-Gl3 levels or both. The genetic variants identified were: p.Asp313Tyr (4 patients), p.Arg220Gln (3 patients) and M290I (1 patient). None of the identified variants is pathogenic.
Conclusions76% of HD Centers of the CAM participated in the study. This is the first publication to describe the prevalence of FD in the HD-population of a region of Spain as well as its average α-GAL-A-activity and plasmatic Lyso-Gl3 levels. It is also the first study that combines a cross-sectional design with a prospective follow-up design. This study has not identified any FD patient.
El objetivo del estudio ha sido realizar un mapa descriptivo de la enfermedad de Fabry (EF) en la Comunidad de Madrid, así como realizar un despistaje de la EF a todo paciente incidente durante un período de 3 años.
Criterios de inclusiónPacientes mayores de 18 años, excluyendo aquellos diagnosticados de cualquier otra enfermedad hereditaria con afectación renal diferente de la EF, que firmaran el consentimiento informado. Criterios de exclusión: pacientes menores de edad, no estar de acuerdo o no ser capaces de firmar el consentimiento informado.
ResultadosSe incluyeron 3.470 pacientes (63% hombres), con una edad media de 67,9±9,7 años, de los cuales 2.357 eran pacientes prevalentes y 1.113 incidentes. En el caso de los pacientes prevalentes, el tiempo medio en hemodiálisis (HD) fue de 45,2 (DE 51,3) meses. En pacientes incidentes, la proteinuria estuvo presente en el 28,4%. No hubo diferencias estadísticamente significativas en la actividad plasmática de alfa-galactosidasa A o valores de Lyso-Gl3 al comparar las poblaciones de incidentes y de prevalentes, y tampoco entre hombres y mujeres. Se realizó estudio genético en 87 pacientes (2,5% de los pacientes): 60 varones con disminución de la actividad enzimática y 27 mujeres con una disminución de la actividad enzimática, aumento de los niveles de Lyso-Gl3 o ambos. Las variantes genéticas identificadas fueron: p.Asp313Tyr (4 pacientes), p.Arg220Gln (3 pacientes) y M290I (un paciente). Ninguna de las variantes identificadas fue patógena.
ConclusionesEl 76% de los centros de HD de la Comunidad de Madrid participaron en el estudio. Este es el primer estudio epidemiológico prospectivo de EF en la población de HD de una región de España. También es la primera vez que se describen niveles de alfa-galactosidasa A y Lyso-Gl3 en HD, sin embargo, en este estudio no se han identificado pacientes con EF.
Fabry disease (FD) is a lysosomal storage disease, with an X-chromosome-linked inheritance, caused by the deficiency of the α-GAL-A lysosomal enzyme due to mutations in its codifying gene.1,2 This enzyme is responsible for the hydrolysis of the terminal alphagalactosyl residues of glycosphingolipids and its deficiency causes their progressive storage, mainly globotriaosylceramide (Gb-3), in multiple cell types and organs.1,2 The storage material also triggers deleterious signaling cascades as inflammatory and fibrotic ones.1–4 These pathways are also activated by globotriaosylsphingosine, Lyso-Gl3,5,6 which is a water-soluble molecule that arises from the deacylation of Gb3 and a proposed biomarker in FD with an important role in diagnosis.7–9
There are two main phenotypic groups: classical and late-onset. In the classic phenotype, there is a severe enzymatic deficiency, and disease manifestations occur already in childhood. Pediatric patients suffer from intense pain crisis in hands and feet, hypohidrosis, cornea verticillata, intermittent fever, gastrointestinal involvement and angiokeratomas. The disease progresses with myocardial, cerebrovascular and renal involvement, resulting in premature death. Late-onset patients develop cardiac and renal signs and symptoms in adulthood, years later in average than classic patients.1,2
Regarding renal involvement, Gb3 is stored globally in the kidney: in podocytes, mesangium, endothelium of the glomerular capillaries, tubular epithelia, interstitial and endothelial cells as well as in the muscular layer of arteries and arterioles. Gb3-deposits per se mediate an array of effects that contribute to FD nephropathy: tubular cell damage, cellular proliferation that will lead to mesangial expansion and vasculopathy, endothelial dysfunction which will also contribute to the vasculopathy, podocyte damage and activation of inflammation and fibrotic signaling pathways.10–12 Fibrotic patterns in FD correspond to glomerulosclerosis and interstitial fibrosis. In average, male patients with the classic phenotype have end renal failure in their forties.1,2
In heterozygous female FD patients, phenotypes can range from asymptomatic as to presenting with similar severity as classic males, depending on the extent of silencing of the X-chromosome carrying the mutated α-GAL-A-gene.1,2,13 In a recent retrospective study in a large cohort of patients, 19% of classic FD female patients had a cardiac, renal or cerebral event before the first visit, in comparison with 30% of classical males.14
The estimated prevalence of FD is of 1:80,000–1:117,000 of live births,15,16 although data from newborn screenings provide a wide range of published prevalences.17–19 The prevalence of FD in patients with renal, cardiac or cerebrovascular disease is much higher than the prevalence in the general populations since in FD these are considered High Risk Populations (HRP). On the other hand, as in other genetic disorders, in FD some “mutations” previously categorized as pathogenic are now considered benign α-GAL-A genetic variants, therefore in older studies prevalence might be overstated. Indeed, Doheny et al. (2017) reviewed the published prevalence of FD in HRPs considering the improved knowledge of FD mutations: of the 23,954 males screened in HD in previous publications, the real prevalence decreased from 0.42% to 0.21%.
FD is one of the few Rare Diseases with an available treatment. Since 2001 enzymatic replacement therapy (ERT) is available and more recently a chaperone therapy has also been approved in Europe.2 Many studies have highlighted the importance of initiating specific FD therapy before irreversible organ-damage has been stablished to improve outcomes.2,20–22
The present study screened for FD in HD patients in the region of Madrid (CAM). The study had two stages, first a cross-sectional design to evaluate HD-prevalent patients, followed by a three-year period prospective design to analyze HD-incident patients.
Materials and methodsStudy designObservational, multicentric, cross-sectional and prospective study. This study screened for FD in the HD-prevalent population in the Comunidad de Madrid (CAM) with 6.642 million inhabitants as well as analyzed the incidence of FD over the period of three years in the incident HD-population. Secondary objectives were to conduct a family study of the new diagnosed cases and to offer genetic information to patients and family members newly diagnosed with FD.
The study was approved by the HD Unit dependent Ethics Committee (October 13th, 2015 (meeting minutes number 17/15), and modified in November 29 2016 (meeting minutes number 20/2016)). All the research was compliant with the individual rights of the patient (Helsinki declaration, Fortaleza act, 2013) and Oviedo convention (1997).
MethodologyInclusion criteria: patients older than 18 years on HD in the CAM, excluding patients diagnosed with any other hereditary disease with renal involvement different from FD, that sign the IC. Exclusion criteria: underaged patients or not agreeing or not being capable of signing the IC.
The screening algorithm is depicted in Fig. 1. α-GAL-A enzyme activity, Lyso-Gl3 levels and genetic analysis was determined in plasma by means of dried blood spots on filter paper. In the study visit the date of sample collection date, age, sex, kidney disease etiology were recorded. Additionally, date of HD initiation on HD-prevalent patients and proteinuria in HD-incident patients was also collected. Diagnostic tests were performed at the medical laboratories of ARCHIMED Life Science GmbH (ARCHIMEDlife; www.archimedlife.com).
Statistical analysis
Statistical analysis for the primary objective and the descriptive of the variables included are detailed below. Quantitative variables will be described with measures of centralization and dispersion (mean, SD, mediana, minimum, maximum, interquartile range). Data was analyzed by SPSS v22. When comparing groups of non-normal distribution have been compared using wilcoxon rank-sum test. Confidence intervals will be calculated at 95%, where necessary, for the result variables associated with the primary and secondary objectives. Statistical significance was considered as a two-sided p-value<0.05.
Results24 HD Units of the CAM participated in the study from the beginning of 2016 to the end of 2019. From a total of 3,470 patients, 2357 HD-prevalent patients and 1,113 HD-incident patients were included in the study. Patients’ characteristics are described in Table 1 and the kidney disease etiology is presented in Fig. 2.

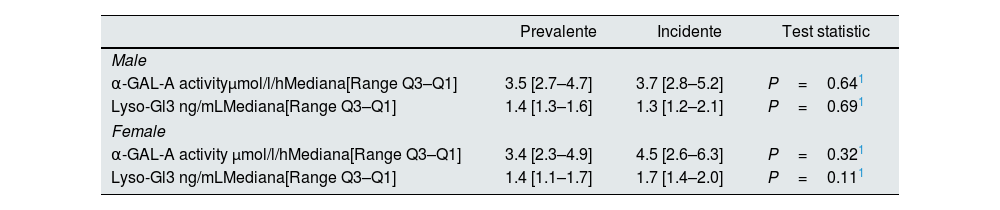
Plasmatic α-GAL-A activity and Lyso-GL-3 values segregated by type of population and sex are shown in Table 2. There were no statistical differences between HD-incident and HD-prevalent populations and neither between males nor females.
Plasmatic α-GAL-A activity and Lyso-Gl3 levels in both populations.
| Prevalente | Incidente | Test statistic | |
|---|---|---|---|
| Male | |||
| α-GAL-A activityμmol/l/hMediana[Range Q3–Q1] | 3.5 [2.7–4.7] | 3.7 [2.8–5.2] | P=0.641 |
| Lyso-Gl3 ng/mLMediana[Range Q3–Q1] | 1.4 [1.3–1.6] | 1.3 [1.2–2.1] | P=0.691 |
| Female | |||
| α-GAL-A activity μmol/l/hMediana[Range Q3–Q1] | 3.4 [2.3–4.9] | 4.5 [2.6–6.3] | P=0.321 |
| Lyso-Gl3 ng/mLMediana[Range Q3–Q1] | 1.4 [1.1–1.7] | 1.7 [1.4–2.0] | P=0.111 |
Cut-off values: α-GAL-A activity>2.8μmol/l/h; Lyso-Gl3: <3.5ng/ml. HD: hemodialysis; SD: standard deviation (95% CI). N is the number of non-missing value.
1Kruskal–Wallis.
A genetic study was performed in 87 patients, which represents 2.5% of the total screened population. 60 male patients with a decreased enzymatic activity were studied. 27 female patients with an altered level of α-GAL-A activity, of Lyso-Gl3 or both were evaluated. No genetic variant was identified in 79 patients (Fig. 3). The genetic variants identified were: p.Asp313Tyr (4 patients), p.Arg220Gln (3 patients) and M290I (1 patient). None of the identified variants is pathogenic.
Discussion
76% of HD Centers of the CAM participated in the study. To our knowledge, this is the first FD epidemiological study with a cross-sectional and prospective follow-up design published in the literature. It is also the first time that the average α-GAL-A-activity and plasmatic Lyso-Gl3 levels of the HD population of the CAM has been described, which will help to stablish more precise cut-off values in future studies, necessary for the more accurate identification of FD patients.
The present study has not identified any FD patient neither in the screened HD-prevalent population nor in the HD-incident one. There are several reasons that could explain why there have been no positive FD patients identified. On one hand, another FD-screening in HD-patients at national level in Spain was started at the same time that our study was being set up in the CAM. The results from this other national study have not yet been published, therefore the data on both the number of screened patients as well as the number of FD-identified patients in the CAM is missing by now. Consequently, there is the possibility that FD-positive patients in the CAM were already identified by this other study23 or by other means, yet these patients were not included in our study.
On the other hand, taking a closer look at the evolution of FD prevalence in HRPs24 over time, in males, the prevalence decreases in all HRPs when comparing data collected in two consecutive periods, 2000–2008 vs. 2009–2017. In HD, the prevalence changed from 0.32% to 0.17%. In another study that analyzed diagnostic delays in FD comparing two consecutive 5-year-periods from 2001 to 2013 in cardiovascular HRPs, the mean time from diagnosis had diminished from 18 to 13 years. Therefore, it is possible that the growing awareness of the disease among health care professionals leads to earlier diagnosis and management of these patients, which would limit the number of unidentified FD patients in the HD subpopulation.24 In females, on the contrary, the proportion of identified FD-patients in later screening increases24 as well as the number of screened females.
Regarding the diagnostic algorithm in women, the present research has used the screening modality of analyzing α-GAL-A and Lyso-Gl3 simultaneously, proceeding to the genetic the analysis in the case one of these parameters were to be abnormal.8 Most diagnostic algorithms recommend directly performing the genetic analysis in women since due to the lyonization process, α-GAL-A plasma activity can be normal in female FD patients.2,20–22 Nevertheless, in a recent study8 with 12,000 screened females following our same algorithm, the highest positive predictive value corresponded to the group with abnormal levels of both Lyso-Gl3 and α-GAL-A activity (97%), followed by abnormal Lyso-Gl3 only (39%) and abnormal α-GAL-A activity only (6%). Indeed, the negative predictive value was of 100% when the levels of Lyso-Gl3 and α-GAL-A activity where within normal ranges, as assessed by sequencing samples from for almost 400 females.
Screening for FD among patients with chronic kidney disease has mostly been done in haemodialysis patients and kidney transplant recipients, for whom the renal damage is already irreversible. Thus, the prevalence of FD in haemodialysis patients could be underestimated.24
Early diagnosis is crucial for correct patient management as well as identification of affected family. In FD, renal function is a predictor of outcomes both in males and females.21,25 Therefore standardizing early diagnosis programs in patients with Red-flag-Fabry symptoms within early stages of CKD and CKDs of unknown etiology would be essential. The impact of such effort would be of greatest value with the leadership and consensus of the Medical Societies In our view, it would be desirable that a FD test was given to any HD-incident patient as a routine clinical practice, yet one of the added values of screening studies is to improve FD awareness in HD units, facilitating a more.
Authors’ contributionsThe main authors study concept, design and execution; article writing and critical revision.
All signing authors read and approved the final manuscript.
Ethics approval and consent to participateThe study was approved by the HD Uni dependent Ethics Committee (October 13th 2015 (meeting minutes number 17/15), and modified in November 29 2016 (meeting minutes number 20/2016)). All the research was compliant with the individual rights of the patient (Helsinki declaration, Fortaleza act, 2013) and Oviedo convention (1997).
Consent for publicationNot applicable.
Availability of data and materialsThe datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
FundingThis work was partially supported by grants from Sanofi-Genzyme (Investigator Initiated Research Grant 2015) given to the Sociedad Madrileña de Nefrología. Funders played no role in the design, collection, analysis, or interpretation of the data or in the decision to submit the manuscript for publication.
Conflict of interestsThe main authors have received support from pharmaceutical companies with treatments for Fabry and other rare diseases.
Our special gratitude towards the patients for their voluntary participation, to the participating centers and to the Sociedad Madrileña de Nefrología for its support and collaboration.



