



Fabry disease is a rare X-linked lysosomal storage disorder of glycosphingolipids, caused by the partial or complete deficiency of the lysosomal enzyme alpha-galactosidase A (a-Gal A). The missense mutation pN215S usually causes a milder form of the disease with isolated cardiac involvement. We report a case of a male Fabry patient with the pN215S mutation and a generalized disease. He suffered a relapse in proteinuria which responded to increased doses of the administered recombinant enzyme. Individualization of enzyme replacement therapy must be considered in selected cases characterized by clinical deterioration.
La enfermedad de Fabry es un trastorno hereditario raro ligado al cromosoma X, que se caracteriza por un almacenamiento lisosómico de glucoesfingolípidos causado por una deficiencia parcial o completa de la enzima lisosómica α-galactosidasa A (a-GalA). La mutación sin sentido pN215S suele provocar una forma más leve de la enfermedad, con afectación cardíaca aislada. Se describe un caso de enfermedad de Fabry en un paciente varón con la mutación pN215S y enfermedad generalizada. El paciente presentó una recidiva de la proteinuria que respondió al aumento de dosis de la enzima recombinante administrada. Debe considerarse la posible conveniencia de una individualización de la terapia sustitutiva enzimática en casos seleccionados que presenten deterioro clínico.
Fabry disease is a rare genetic lysosomal storage disorder of glycosphingolipids with X-linked transmission and an estimated incidence of 1:40,000–1:117,000 live male births.1 Partial or complete deficiency of the enzyme alpha-galactosidase A (a-Gal A) results in altered metabolism and progressive lysosomal accumulation of the substrate (mostly globotriaosylceramide, Gb3).2 The responsible gene is located on the long arm of the chromosome X (Xq22). More than 600 mutations have been identified with variable phenotypical expression.3
Clinically we distinguish the classical form and two variants, cardiac and renal. In the classical form clinical manifestations appear during childhood or early adolescence including acroparesthesias, angiokeratomas and corneal opacities.4 Progressive accumulation of Gb3 in the kidneys, heart and central nervous system lead to renal failure, hypertrophic cardiomyopathy and cerebral vascular accidents limiting life expectancy. The cardiac variant of the disease is associated with residual alpha-galactosidase A activity (>1%), appearing later in life. The patients suffer from left ventricular hypertrophy and hypertrophic cardiomyopathy with decreased systolic function. Other cardiac manifestations include valvular disease with thickened valves (especially left-sided) and regurgitation, myocardial ischemia, arrhythmias (frequently supraventricular) and ECG changes such as voltage criteria for LVH and repolarization abnormalities, shortened P-R, A-V block or bundle branch block.5 The clinical picture is dominated by the aforementioned cardiac manifestations, while the classical signs and symptoms are usually absent. Some patients may present a degree of proteinuria without severe renal failure.6
Advances in the application of molecular genetic techniques have enabled the development of directed protein therapies for lysosomal storage diseases. In case of Fabry disease two formulations of recombinanat enzyme are currently available. Agalsidase-alfa (Replagal®, Shire) is derived from human skin fibroblasts and is administered intravenously at dose of 0.2mg/kg every 14 days. Accordingly, agalsidase-beta (Fabrazyme®, Genzyme) is produced by Chinese Hamster ovary cell line and is given intravenously at dose of 1mg/kg every 14 days.
We present the case of a male Fabry patient who was diagnosed with the missense mutation pN215S who in addition to cardiac involvement also presented serious extracardiac clinical manifestations. In our case, twenty months after the initiation of agalsidase-alfa in the conventional dose, an increment of proteinuria and left ventricular mass were noted. Switching the patient to double dose of the enzyme led to reduction of proteinuria and reestablishment of the cardiac indexes.
Case reportA 41-year old male was presented in the Renal Outpatient Clinic with proteinuria of 0.5g/d and left ventricular hypertrophy. His renal function was normal (eGFR 122ml/min/1.73m2). The patient underwent a renal biopsy which showed glomeruli with enlarged podocytes displaying abundant fine, granular and lucent protoplasm. Some tubular epithelial cells had a vacuolated and lucent cytoplasm or atrophy while mild interstitial fibrosis was also present (<10%). Immunofluorescence showed IgM deposition in mesangium of two glomeruli with granular distribution. Histological findings in association with the clinical manifestations led to further investigation toward diagnosis of Fabry disease. Indeed, low activity of α-Gal A in plasma and in leucocytes (0.3nmoles/ml/h and 1.5nmoles/mg protein/h, respectively) led to the aforementioned diagnosis. The subsequent familiar genetic investigation revealed that both he and his mother, a 70-year old female, had the pN215S missense mutation, corresponding to adenine replacement in the position 10135 with guanine (10135A→G).
Monitoring of the patients, performed twice a year, included biochemical exams of renal function and 24-h measurement of proteinuria, cardiac ultrasound and pro-BNP levels, plasma and urine levels of Gb3 as well as the titer of the anti-agalsidase antibodies and brain MRI scan every other year. The female suffered from left ventricular hypertrophy, without renal or CNS involvement. On the contrary, male patient had a complex phenotype with left ventricular hypertrophy, proteinuria and CNS lesions. Brain imaging with MRI scan showed vascular dolichoectasia in the vertebrobasilar junction and punctuated white matter lesions in the frontal lobe as well as in the basal ganglia.
The male patient was administered agalsidase-alfa (Replagal®, Shire) in the recommended dose of 0.2mg/kg/d every 2 weeks and an ACE inhibitor in the maximum tolerated dose (ramipril 5mg daily). Enzyme replacement therapy began 3 months after the histologic diagnosis.
During the first 20 months both renal and cardiac indexes remained stable and no other relevant event was noticed. At that point in the male patient a steep increase of proteinuria (from 560 to 2536mg/d) as well as an increase in left ventricular mass (from 412 to 464.5g) and pro-BNP levels (from 190 to 266.5pg/ml) were observed. After measurement of Gb3 levels in plasma (5.86nmol/ml) and exclusion of development of neutralizing anti-agalsidase-a IgG and IgA antibodies we considered to adapt the therapeutic regimen. Thus we administered, with the same frequency, double dose of enzyme (0.4mg/kg/d). Seven months later, proteinuria decreased from 2536 to 986mg/d and the indexes of cardiac size were reestablished. Accordingly plasmatic levels of Gb3 were 5.11nmol/ml. Targeting to verify this apparent dose-dependent effect, the conventional dose was re-administered and one month later proteinuria relapsed (2886mg/d) (Fig. 1). Therefore the patient switched to the double dose. After 24 months, he still presents albuminuria of 1053mg/d and stable cardiac size and function (LV mass=414.8g, pro-BNP=166.5pg/ml). Repeated brain MRI scan showed no evolution of the lesions.
Discussion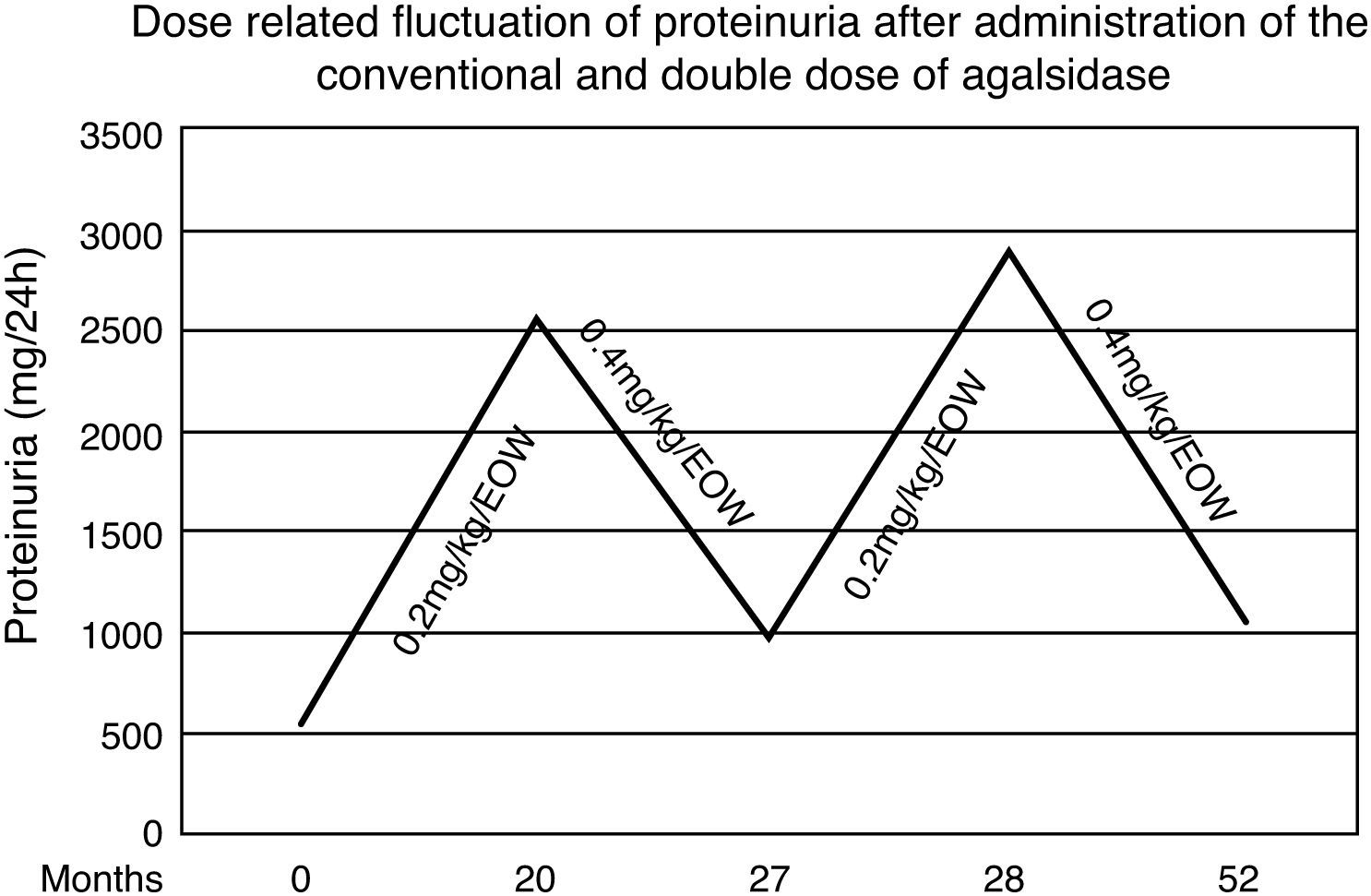
Although, all types of renal cells as well as the endothelium of the blood vessels may be affected, podocyte injury is central in the pathophysiology of Fabry nephropathy.7 Progressive damage of the glomerular filtration barrier leads to the early appearance of the albumin in the urine sub form of microalbuminuria.8 Non-nephrotic range proteinuria developes frequently during the 3rd decade of life and renal failure in the 4th–5th decade, progressing to end stage renal disease after 1–13 years.9 In Fabry patients proteinuria is an independent factor of worsening of renal function and progression to ESRD. Retrospective studies of ERT-untreated cohorts showed that the level of proteinuria was independently associated with the rate of decline of eGFR.10,11 Administration of ERT improves renal histopathology leading to significant clearance of the Gb3 deposits from the renal interstitial vascular endothelium and other glomerular cells.12,13 Clinically, the maximal renal protection is achieved when ERT is given before the development or in an early stage of CKD (eGFR >60ml/min/1.73m2 or proteinuria <1g/d).13–15 On the contrary, the effectiveness of ERT is much less consistent in face of an advanced renal failure and irreversible organ damage.16,17 Thus proteinuria must be a major therapeutic target in Fabry nephropathy.
Our patient was diagnosed with the missense mutation pN215S (transition of A→G in codon 215 of exon 5 with substitution of an asparagine by a serine). This genetic defect usually leads to a late-onset phenotype, dominated by cardiac manifestations.18,19 However, in this case the phenotypic expression was different with renal and CNS involvement besides cardiac hypertrophy. To our knowledge this is the first case of a Fabry patient with the pN215S mutation and extracardiac clinical manifestations.
Although the patient had normal renal function, control of proteinuria to levels <1g/d was necessary. Low blood pressure (100/65mmHg) prevented us from further titration of the ACE inhibitor. Thus, we decided to increase the administered enzyme by doubling the recommended dose. This choice was based upon previous in vitro observation that administration of recombinant human galactosidase is associated with increased tissue enzymatic activity in a dose-dependent manner.20 During the next seven months we observed a gradual decrease in proteinuria levels as well as in left ventricular mass. This dose-dependent effect was verified by switching the patient to the conventional dose of agalsidase-alfa for one month period, after which a re-deterioration of proteinuria was noticed. Therefore the patient switched to the double dose regaining the prior albuminuria levels and stable cardiac size and function. The findings sustained 24 months later.
Although long-term administration of enzyme replacement therapy (ERT) appears safe and effective leading to clinical stabilization,21 there are still controversies regarding the appropriate ERT regimen in case of progressive clinical deterioration. A dose-dependent effect has been observed in such a case with advanced nephropathy.22 In an open-label, prospective clinical trial of 11 male patients with advanced Fabry nephropathy (mean baseline eGFR 53.7±6.3ml/min/1.73m2) and rapid annual decline of eGFR (−8±0.8ml/min/1.73m2), switching agalsidase-alfa regimen from the conventional biweekly dosing to weekly infusions of 0.2mg/kg for a 24-month period slowed eGFR's decline (−3.3±1.4ml/min/1.73m2).23
The question of the optimal ERT dose has also been tested by Vedder et al. who administered 3 different ERT regimens in 52 Fabry patients (0.2mg/kg agalsidase-alfa, 0.2mg/kg or 1mg/kg agalsidase beta). After 12 months of follow-up the patients who received 1mg/kg of agalsidase-beta presented decreased levels of urinary Gb3 levels, independent of their anti-agalsidase antibody status. Moreover, a reduction of left ventricular mass was observed.24 However, the results of a comparative trial between the two products administered in 34 patients at the same dose (0.2mg/kg biweekly), showed no difference regarding reduction of left ventricular mass after 12 and 24 months of treatment. There was also no difference concerning other clinical or laboratory parameters (eGFR, pain, plasma and urinary Gb3 levels, anti-agalsidase antibodies) or treatment failure between the two treatment groups.25 In our case administration of double of the recommended dose of agalsidase-alfa, besides better compliance, was followed by a significant decrement of proteinuria as well as decrease of the left ventricular mass.
In conclusion, individualization of ERT should be considered in selected cases of Fabry disease when conventional dosing and symptomatic treatment are not effective to control clinical manifestations. However, additional investigation is required in order to evaluate cost–effectiveness of such an intervention.
Conflict of interestThe authors declare no conflict of interest.



