




Uno de los retos a los que debe enfrentarse la nefrología moderna es el de identificar biomarcadores que se asocien a patrones anatomopatológicos o a mecanismos patogénicos definidos y permitan el diagnóstico no invasivo de la causa del síndrome nefrótico o establecer subgrupos pronósticos en cada tipo de enfermedad, prediciendo la respuesta al tratamiento y/o la aparición de recidivas. Los avances en el conocimiento de la patogenia de las distintas enfermedades causantes de síndrome nefrótico, sumados al progresivo desarrollo y estandarización de las técnicas de proteómica plasmática y urinaria, han permitido ir identificando un número creciente de moléculas que podrían ser útiles para los fines anteriormente mencionados. En el momento actual, los datos de muchos de los candidatos identificados, sobre todo mediante técnicas de proteómica, son todavía muy preliminares. En la presente revisión, se resume la evidencia disponible sobre las moléculas que en la actualidad cuentan con mayor evaluación en estudios clínicos.
One of the major challenges modern nephrology should face is the identification of biomarkers that are associated with histopathological patterns or defined pathogenic mechanisms that might aid in the non-invasive diagnosis of the causes of nephrotic syndrome, or in establishing prognosis sub-groups based on each type of disease, thus predicting response to treatment and/or recurrence. Advancements in the understanding of the pathogenesis of the different diseases that cause nephrotic syndrome, along with the progressive development and standardisation of plasma and urine proteomics techniques, have facilitated the identification of a growing number of molecules that might be useful for these objectives. Currently, the available information for many of the possible candidates identified to date, above all those discovered using proteomics, are still very preliminary. In this review, we summarise the available evidence for the different molecules that have been best assessed using clinical studies.
INTRODUCTION
Nephrotic syndrome is defined by proteinuria >3.5g/day in adults and 40mg/m2 in children, associated with hypoalbuminaemia, oedema, hyperlipidaemia, and hypercoagulability.1 The common mechanism in all renal diseases that cause this syndrome is the loss of selectivity of the glomerular filtration barrier, which allows the massive flow of proteins into the urinary space.2 The primary forms are defined as those in which it is not possible to establish a systemic disease responsible for this condition. The secondary forms include renal lesions that appear as a consequence of other diseases that are accompanied by characteristic extra-renal signs and symptoms. The histopathological lesions that are most commonly responsible for nephrotic syndrome are minimal change nephropathy (MCN), focal segmental glomerulosclerosis (FSG), membranous nephropathy (MN), and less frequently, membranoproliferative glomerulonephritis (MPG), as a primary glomerulopathy, and diabetic nephropathy and immunoglobulin deposit nephropathies as secondary nephropathies.1-3 In children (due to the marked predominance of MCN4,5) and in some secondary forms in adults, it is possible to have a certain level of clinical suspicion regarding what type of histopathological lesion is causing the nephrotic syndrome. However, in the vast majority of cases of adult nephrotic syndrome, it is necessary to perform a renal biopsy in order to reach a reliable diagnosis, establish a prognosis, and choose the most appropriate treatment. Without a doubt, one of the pending challenges that modern nephrologists face is the identification of biological markers that are associated with defined pathogenic mechanisms or histopathological patterns and that allow for a non-invasive process of diagnosing the cause of the nephrotic syndrome or establish prognostic sub-groups in each type of disease, thus predicting the response to treatment and/or recurrences.
The continuous advancements made in our understanding of the pathogenesis of different diseases that cause nephrotic syndrome, together with the progressive development and standardisation of blood and urine proteomic techniques, have facilitated the identification of a growing number of molecules that could be useful for the aforementioned goals if sufficient sensitivity and specificity were shown for identifying the type of renal damage and/or the response to treatment or prognosis of the disease. Currently, the information regarding many of the candidates identified until now, above all those using proteomic techniques, are still very preliminary. In this review, we discuss the available evidence regarding the different molecules that have been most comprehensively evaluated in clinical studies.
BIOMARKERS IN MINIMAL CHANGE NEPHROPATHY
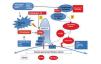
MCN is characterised by an absence of lesions visible using optical microscopic techniques, as well as an absence of deposits in immunofluorescence studies.1,2 The only demonstrable lesion in this disease is the fusion of podocyte foot processes revealed using electron microscopy. The common association with atopy, infections, vaccinations, and lymphoproliferative processes, along with the fact that the majority of these patients respond to treatment with steroids, immunosuppressants, and immunomodulators, have served as solid evidence to suggest the participation of the immunological system in its pathogenesis. In 1974, Shalhoub suggested that the damage to the filtration barrier could be due to the production of lymphokine produced by T-lymphocytes.6 Since then, several studies have demonstrated the existence of deregulations of the immune response, especially in T-cells, and have suggested that MCN could be due to a primary alteration in the function of these cells.7-11 In addition, a predominantly Th2 response has been identified in the active phase of the disease.12 Koyama et al.13 developed a T-cell hybridoma capable of inducing proteinuria by modifying the electric charge of the filtration barrier, and other authors have isolated monocyte proteins and soluble proteins associated with the activation of the immune system, produced by regulatory T-lymphocytes, capable of inducing proteinuria without altering the electric charge of the filtration barrier. The recent evidence involving the response to rituximab of patients dependent on steroids and calcineurin inhibitors suggests that B-lymphocytes could play a relevant role in many patients, whether directly or in cooperation with T-lymphocytes.14 Despite the clear evidence implicating the immunological system in the pathogenesis of MCN, we have yet to identify the mediators or mechanisms that cause the podocyte lesions. However, studies in recent years have provided novel results regarding molecules that might be useful as biomarkers for the diagnosis and monitoring of activity/response to treatment of this disease (Figure and Table 1).
Urine levels and podocyte expression of CD80 (B7.1)
Recent studies have indicated that podocyte cells in certain circumstances can acquire the phenotype and/or function of dendritic cells and can be induced to express CD80 (B7.1).15,16 CD80 is a transmembrane protein expressed in antigen-presenting cells that, when joined to its ligand, CD28, which is present in T-lymphocytes, produces a co-stimulation signal for these molecules, which is essential for lymphocyte activation.18 Normal podocytes do not express CD80. It has been shown in experimental models that the expression of CD80 in podocytes is associated with the appearance of nephrotic protienuria.16 Currently, we do not know the functional significance of the neo-expression of CD80 by the podocyte, and it has not been shown whether this is related to the alterations in filtration membrane that produce proteinuria. The expression of CD80 can be induced by oxidative stress or following stimulation with lipopolysaccharide (LPS),16 through signalling processes mediated by Toll-like-3 receptors18 and interleukin (IL) 13,19 and this expression is not dependent on lymphocytes, since it has also been induced in knock-out animals models without T-lymphocytes.16 The expression of CD80 in podocytes has also been demonstrated in biopsies of humans with MCN, and it has recently been described that CD80 levels are elevated in MCN patients during the initial breakout but normalise after remission, but are not found in high levels in other nephropathies that cause nephrotic syndrome, such as MN or FSG, nor in other types of glomerular disease.20 The available clinical results are still preliminary, but they appear to indicate that measuring urine CD80 could be useful for the non-invasive diagnosis of MCN and for a differential diagnosis between MCN and FSG and/or to monitor the activity of the disease.
Interleukin 13
From the association between MCN and Hodgkin’s disease, along with the evidence that IL-13 is an autocrine growth factor for Reed-Stemberg cells,22-24 substantial experimental (and to a quite lesser degree, clinical) evidence has been produced that correlates IL-13 with the induction of structural changes in the podocyte that are capable of altering the selectivity of filtration and causing nephrotic syndrome. Recently, it has been shown that the expression of the IL-13 gene is elevated both in CD4 and CD8 lymphocytes in children with corticosteroid-sensitive nephrotic syndrome during recurrences.12 This increase is associated with higher levels of IL-13 in the cytoplasm of T-cells25 and downregulation of IL-8 and IL-12 proinflammatory cytokines in monocytes.26 A correlation has also been described between polymorphisms at the 3' untranslated region of the IL-13 gene and the clinical progression of MCN. The expression of IL-13 messenger ribonucleic acid (mRNA) in mononuclear cells in AAT haplotype patients, which is associated with multiple recurrences, is significantly greater than in patients with the GCC haplotype, which is associated with long-term remission.27 Recently, it has also been shown that IL-13 receptors are present in podocytes, and the stimulation of cultured podocytes with IL-13 induces functional changes consisting of a decreased transepithelial electrical resistance and STAT6 phosphorylation.28
A transgenic mouse model has also been developed for IL-13.29 In this model, the transgenic mice over-express and have permanently high circulating levels of IL-13, but not so for other Th1 cytokines (IL-2, interferon [IFN]) or Th2 cytokines (IL-4), and develop nephrotic syndrome with ultrastructural renal lesions identical to those observed in MCN. Studies of gene expression as well as those involving immunofluorescence show a reduced expression of nephrin, podocin, and dystroglycans, and an increase in podocyte expression of CD80. In previous experimental studies, it had already been shown that IL-13 in combination with IL-1 and IFN-gamma could induce the expression of CD80 in proximal tubular epithelial cells, but not in podocytes. The evidence that IL-13 can induce the expression of CD80 in podocytes and that this is associated with the appearance of nephrotic syndrome establishes a very interesting link between these processes, as this opens the possibility for considering that certain MCN patients could have proteinuria caused by the direct effects of IL-13 on the tertiary structure of the podocyte, and at the same time, generates the hypothesis regarding the possible clinical usefulness of studying the IL-13–CD80 pathway in MCN patients, in relation to the clinical progression, response to treatment, and prognosis of this disease. In addition, the evidence that IL-13 plays a key role in the production of IgE and IgG4 in nephrotic patients, in contrast to what occurs in asthma patients, in which the production of IgE is primarily dependent on IL-4,30 could contribute to explaining the relationship between MCN and atopy.
Serum levels and protease activity of circulating haemopexin
Plasma haemopexin (Hx) is a ß-1 glycoprotein whose molecular weight varies based on the level of glycosylation.31,32 In addition to its basic function, which consists of binding and transporting the free haeme group and maintaining iron homeostasis, Hx plays a role as an antioxidant.33 Hx is considered as an acute phase reactant, since its synthesis in the liver increases in response to IL-6 and IL-1.34,35 Several circulating isoforms of Hx exist that have yet to be well described. However, it is held that Hx circulates in the bloodstream in an inactive form in healthy patients. From normal human plasma, an isoform of Hx has been identified with protease activity that can be inhibited in vitro using various inhibitors of serine protease or using ATP.36,37 This isoform is capable of inducing glomerular lesions similar to those observed in MCN in both in vitro renal tissue and after intra-renal infusions in in vivo rats. The induction of proteinuria and podocyte effacement is associated with reduced expression of ecto-apyrase and podocyte retraction.38,39
Measuring serum and urine levels and protease activity of Hx indicates that patients with MCN in the active phase have reduced levels of circulating Hx and increased protease activity. In urine samples taken from patients with MCN in the breakout phase, and in contrast to the pattern in other diseases produced by nephrotic syndrome, an isoform of Hx with increased protease activity circulates, although the clinical significance of this phenomenon is unknown40. More studies are needed to determine whether these results are specific to MCN and if they provide any value in the diagnosis or monitoring of patients.
Serum levels of soluble interleukin 2 receptor
The IL-2 membrane receptor is a protein composed of three chains: alpha, beta, and gamma.41 Unstimulated T-lymphocytes express the beta and gamma sub-units. After the T-lymphocyte is activated by recognising an antigen through the T-cell receptor (TCR), in association with co-stimulatory factors, the alpha chain is expressed, which, together with the other two chains, forms the functionally active membrane receptor for IL-2 (IL-2R). In response to the TCR stimulation, the T-lymphocyte produces IL-2, which causes the clonal expansion and activation of T-lymphocytes through binding to the membrane receptor (IL-2R). Due to reasons that are still not completely understood, a soluble form of the receptor (sIL-2R) is generated from the proteolytic rupture of the alpha sub-unit and released into the bloodstream in parallel and proportional to the expression of the membrane IL-2R. The exact function of the soluble receptor is unknown. It is believed that it might be capable of capturing circulating IL-2 molecules, thus modulating the quantity of this cytokine that can be joined to the cellular receptor. The circulating levels of sIL-2R are considered to be an indirect measurement of T-cell activation.42,43 Substantial evidence indicate that, during the acute phase of MCN, patients have very high levels of sIL-2R that then return to normal after remission.44-52 However, elevated levels of sIL-2R have also been described in patients with various types of inflammatory and immunological diseases, in other primary nephropathies that cause nephrotic syndrome, and lupus nephropathy, and until now, no studies have been carried out to analyse the sensitivity or specificity of this parameter, and so it is not possible to conclude whether measuring circulating levels of sIL-2R could provide some diagnostic or prognostic value as compared to conventional methods.
ABCB1 and glycoprotein-P
Glycoprotein-P (CD243) is a transmembrane protein belonging to the family of ATP-binding cassette transporters (ABC-transporters), whose synthesis is coded for by the ABCB1 gene (previously referred to as MDR1), located on the 7p region of chromosome 21. This protein plays a role in the natural detoxification process expressed in several different types of normal human tissues, associated with secretory or barrier functions, and acts as a membrane transport protein responsible for removing drugs and toxins from the cell that have a molecular weight of 300-2000Da, such as xenobiotics, vinca alkaloids, verapamil, and corticosteroids, among others. Glycoprotein-P appears to have two types of activity, protecting the cell from the effects of drugs while also inducing resistance to their activity.53-55 The overexpression of glycoprotein-P is considered as one of the mechanisms that produces resistance to chemotherapy in cancer patients,55 and it has also been suggested that this phenomenon is implicated in the resistance to steroids in autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis.56-58 Recently, it has been shown that IL-2 can induce an increase in the expression of ABCB1 and glycoprotein-P through translocation of the specific transcription factor Y-box protein-1 from the cytoplasm to the nucleus in lymphocytes.59 Through this mechanism, IL-2 could contribute not only to the pathogenesis of minimal change disease, but also to the development of resistance to steroid treatment, above all in patients that, due to multiple outbreaks, have been repeatedly exposed for long periods of time to IL-2 and steroids.59
In a recent study52 in which circulating levels of sIL-2R were measured and the expression of ABCB1 was quantified in the lymphocytes of patients with nephrotic syndrome secondary to MCN, these patients had higher sIL-2R and ABCB1 levels than in healthy control subjects, both during the outbreak and after remission. During the outbreak phase, both sIL-2R and ABCB1 levels were significantly higher in patients with resistance to corticosteroid treatment and those with multiple outbreaks than in patients at their first outbreak or that responded to corticosteroids. After receiving steroid treatment, sIL-2R and ABCB1 levels did not change significantly in patients that were resistant to corticosteroids. In patients that did respond to corticosteroid treatment, sIL-2R and ABCB1 levels decreased significantly following remission, but patients with recurrent forms continued to have significantly higher levels than healthy controls. These data coincide with the results published previously by other authors,59-61 and suggest that, in patients with multiple recurrences that require repeated and prolonged treatment with steroids, exposure to the drug and the persistent activation of T-lymphocytes could be implicated in the development of resistance to corticosteroids or necessitate growing steroid doses in order to induce the same pharmacological effect. The ground for this hypothesis is still quite deficient, and larger confirmatory studies are needed. Even so, this field of research is of substantial potential interest since further elucidation of these relationships could be useful for predicting the response to steroid treatment, the risk of recurrence, and/or early indications for other treatment options based on sIL-2R and ABCB1 levels at the time of diagnosis, or the evolution of these levels during follow-up.
BIOMARKERS IN FOCAL SEGMENTAL GLOMERULOSCLEROSIS
The term “focal segmental glomerulosclerosis” is used to define an entity that presents a defined pattern observable in light microscope analysis, but that has several possible aetiologies and pathogenies.62 FSG is classified as primary or secondary based on whether an aetiology responsible for the condition is identified or not.63 Distinguishing between primary and secondary forms is of paramount interest for both the treatment and prognosis of the condition, since only patients with primary forms that develop nephrotic syndrome and that are not produced by mutations in podocyte proteins are candidates for immunosuppressant or immunomodulating treatment.64 Currently, the differentiation between the two forms is based on the clinical profile of the patients and an ultrastructural renal analysis through electron microscopy. Although it is not an infallible criterion, it is generally considered that primary forms are characterised by nephrotic syndrome and generalised effacement of the podocyte foot processes as observed in electron microscope analyses. In secondary forms, nephrotic-range proteinuria can be observed, but nephrotic syndrome is uncommon and an electron microscope analysis facilitates the identification of a focal segmental distribution of the effacement of podocyte foot process, rather than diffuse.65
The pathogenic mechanisms that produce irreversible damage in the podocytes are poorly understood both in secondary and primary forms, and in the latter group, it is highly probable that no single pathogenic mechanism is shared by all cases.
In recent years, molecular biological techniques have allowed for describing a growing number of mutations that affect structural proteins in the podocyte or proteins involved in the slit diaphragm that could cause the lesions observed in FSG.66,67 In the majority of cases, FSG starts in infancy or adolescence, usually associated with a family history that can take on a myriad of patterns, and in some cases, extra-renal syndromes that facilitate a diagnostic suspicion that the structural renal lesion is due to a mutation in a podocyte protein. However, although it is an uncommon phenomenon, there is evidence for sporadic mutations in the absence of a family history or associated extra-renal symptoms in patients whose FSG starts in adulthood.67 This evidence indicates that, although quite uncommon, some forms of the condition that were previously considered to be primary are in fact due to podocyte protein mutations.
The fact that some patients with primary FSG respond to treatment with corticosteroids and/or immunosuppressants64,65 has led to the belief that, in some cases, the pathogenesis could be related to the activation of the inflammatory and/or immune response, but autoimmune phenomena or deregulations of the immune response with a pathogenic significance have not been demonstrated. The absence of immune deposits in biopsies, the evidence of recurrences after kidney transplantation that respond to treatment with plasmapheresis, immunoabsorption, or lipid apheresis,68-70 and the evidence of transmitting nephrotic syndrome from FSG mothers to newborns71 have all provided a rational basis for generating the hypothesis of a circulating factor or permeability factor (PF) capable of damaging the podocytes.27-29 The existence of this factor (or factors) was purely speculative until it was demonstrated that the plasma of certain FSG patients could produce alterations in the permeability of proteins in in vitro glomeruli.72 From this point, the pathogenic character of PF still has yet to be unquestionably proven in patients with primary forms of FSG. In cases in which molecules characteristic of PF have been identified, no convincing association has been established with the response to treatment or with recurrence after kidney transplantation.
Circulating levels of soluble urokinase-type plasminogen activator receptor
Very recently, a potentially relevant advancement came about when it was found that serum levels of the soluble urokinase-type plasminogen activator receptor (suPAR) is elevated in patients with primary FSG, but not in patients with other glomerular diseases.73 The urokinase-type plasminogen activator receptor (uPAR) is a glycophosphatidylinositol capable of transmitting intracellular signals by binding to membrane integrins.74,75 Its exact function is unknown, but in experimental models is has been shown that the induction of signalling through uPAR in podocytes produces podocyte fusion and proteinuria through a mechanism that depends on the activation of alpha-V beta-3 integrin.76 For unknown reasons, uPAR can be released from the plasma membrane in a soluble form (suPAR).74,75 suPAR has a molecular weight of 20-50kDa, similar to the size predicted for the hypothetical PF described in previous studies.77 Under normal conditions, the concentrations of this molecule are low, but they may become elevated in patients with certain malignant types of cancer, as well as in patients affected with human immunodeficiency virus.78,79 The available evidence indicates that approximately two-thirds of patients with primary FSG have elevated levels of suPAR. In FSG patients that receive a kidney transplant, the presence of elevated levels of suPAR prior to transplantation appears to increase the risk of recurrence of disease in the transplanted kidney, and preliminary evidence suggests that treatment with plasmapheresis may significantly reduce these levels and induce remission.73 At the experimental level, FSG lesions have been induced in transgenic mice that over-express suPAR.73 Experimental results in the same group that described the increased circulating levels of suPAR in patients with primary FSG indicate that suPAR could act through binding to the podocyte β3 integrin, one of the principal proteins that acts to anchor the podocytes to the glomerular basement membrane. The suPAR-β3 integrin junction would cause the activation of the podocyte and produce changes in its structure and function, which would in turn alter the permeability of the glomerular filtration membrane. Although identifying the relationship between increased suPAR levels and FSG was a major breakthrough, since it was the first time in which it was possible to establish an apparently consistent relationship between a circulating factor and the induction of podocyte lesions, it is still unknown which cells release these molecules into the bloodstream, which factors regulate their synthesis, or for what reasons do suPAR levels increase at a certain point, triggering nephrotic syndrome. On the other hand, we must point out that a high percentage of FSG patients do not have elevated circulating levels of suPAR. It has been suggested that, in these cases, the podocyte damage may be produced by signalling through the local uPAR; however, it is also possible that the damage may be caused by pathogenic mechanisms completely unrelated to this pathway. For these reasons, in order to understand the clinical value of suPAR as a possible biomarker for FSG, further studies are needed with larger patient sample sizes and prospective monitoring in order to determine what levels have a diagnostic value and whether the presence of elevated levels of suPAR are correlated with the clinical presentation, response to treatment, or prognosis of the disease. If confirmed, this hypothesis will open a new branch of study and perhaps even reorientate the treatment strategies for FSG patients, while producing a new treatment target for novel interventions that might be capable of reducing suPAR levels or blocking the binding of suPAR and β3 integrin.
Biomarkers, differential diagnosis between minimal change disease and focal segmental glomerulosclerosis, and predicting the response to steroid treatment
In idiopathic nephrotic syndrome caused by MCN or FSG, the response to steroid treatment has been identified as the main long-term prognostic variable, regardless even of the histological substrate, both in children and in adults.64,65 While it is true that resistance to corticosteroids is associated with a greater frequency to a FSG-type histological pattern, many patients with this renal pathology respond to steroids or other immunosuppressant therapies, and the response to treatment significantly improves their prognosis.64 On the other hand, although the majority of MCN patients do respond to steroid treatment, certain patients with unquestionably MCN-type histological lesions are resistant to corticosteroids, whether during the initial phase or over the course of disease progression. This absence of a consistent correlation between the clinical presentation or histological type of lesion and response to treatment has led to several studies orientated towards seeking out new parameters that will facilitate identifying patients based on response to steroid treatment from the moment of diagnosis. Currently, no single biomarker exists that fits this description, but several potential candidates have been recently described. At the histological level, the increase in podocyte expression of CD8019,20 and reduced expression of α-dystroglycans80,81 allows for differentiating MCN from FSG, but the response to steroids has not been established in association with defined patient profiles. Urine levels of CD8020 and TGF-β82 have also been proposed as candidates for differentiating between these two pathologies, but in this case too, no clear association with response to treatment has been demonstrated. In addition to the relationship between the expression of ABCB1/glycoprotein-P and resistance to corticosteroids,52 a possible relationship has also been described between certain polymorphisms of the genes that code for the synthesis of IL-6, IL-4, and TNF-α and the response to steroid treatment in children with idiopathic nephrotic syndrome.83 In addition, certain urine proteomic profiles have been described that vary based on response to steroids, but this parameter has still yet to be evaluated in clinical studies.84,85 In a very recent study,86 the presence of a 13.8kD fragment of 1-B glycoprotein was described in the urine samples of 36% of patients with resistance to corticosteroids, and in no one of patients which were susceptible to corticosteroid treatment. However, this 1-B glycoprotein fragment is also associated with lower glomerular filtration rates, and so the absence of a response could be explained by the fact that this molecule is an indicator of more advanced stages of disease. Although these data are of great interest, they require confirmation in larger clinical studies.
BIOMARKERS IN MEMBRANOUS NEPHROPATHY
New antibodies in primary membranous nephropathy. Diagnostic value, relationship with clinical activity and response to treatment
MN is the primary cause of idiopathic nephrotic syndrome in adults. Its pathophysiology is based on the formation of immune deposits in the subepithelial space, between the lamina rara externa of the glomerular basement membrane and the podocyte. The available evidence indicates that the deposits are formed in situ at the base of the podocyte processes, which then become detached, and the deposits become then fixed to the external edge of the glomerular basement membrane.87,88 The currently accepted pathogenic model of MN is based on its close similarity to Heymann’s experimental nephritis model.89-92 Heymann’s findings in nephritis were never reproduced in humans,93,94 but the evidence provided by this model was the basis for carrying out studies to identify the antigen(s) implicated in the pathogenesis of human MN.
Until very recently, the only evidence that associated the presence of an auto-antibody directed against a podocyte antigen with the appearance of nephrotic syndrome was in neonatal MN that occurs in children of mothers with deficits for the podocyte antigen called neutral endopeptidase (NEP).95 When these mothers have been immunised against the antigen in previous pregnancies, in later pregnancies, anti-NEP antibodies in the maternal blood pass into foetal circulation through the placental barrier, and after the antigen embeds in the foetal podocytes, this forms immunocomplexes and produces proteinuria.
Very recently, by exposing homogenised healthy renal tissue to antibodies from patients with primary MN using western blot techniques, followed by isolation and identification of the antigen, the M-type phospholipase A2 receptor (PLA2R) was identified as the first target podocyte antigen in the autoimmune response involved in primary MN.96
PLA2R has a structural organisation similar to the mannose receptor in macrophages and forms part of a group of membrane receptors belonging to the C-type lectin superfamily. The human receptor was originally cloned from renal tissue, where the podocytes express this molecule at a high level.97 The extracellular domain is large, composed of an N-terminal region rich in cysteine, a type II fibronectin domain, and a region of 8-10 AA for recognising different carbohydrates. It is believed that PLA2R transmits intracellular signals after binding to one or more of the soluble A2 phospholipases.97-99 In experimental models,100 this molecule has been demonstrated to play an important role in the endotoxic shock induced by LPS, since rats that do not have the receptor have a greater resistance to the action of LPS. In humans, its function is unknown. The presence of anti-PLA2R antibodies is considered to be specific to primary MN. The available study results to date (Table 2) indicate that 60%-70% of patients with primary MN have elevated levels of these antibodies.96,101-103
In addition, a clear correlation has been described between antibody titres (especially IgG4) and the clinical activity of the disease,102,103 and treatment with rituximab has been shown to be capable of reducing the antibody titres, in parallel to the reduction in urine excretion of proteins.104 In addition, immunocomplex deposits containing anti-PLA2R antibodies have been identified in the external edge of the glomerular basement membrane using immunofluorescence studies, even in patients in which circulating levels of this antibody are negative, which would indicate that a negative antibody titre (at least using currently available techniques) does not necessarily allow for excluding the diagnosis of MN.105 In recipients of kidney transplants, the presence of anti-PLA2R antibodies could also be of great importance for differentiating between a relapse of MN and de novo post-transplant MN.106 The role of anti-PLA2R antibodies in the pathogenesis of MN is unknown. It has been suggested that the lesion could be produced after the formation of immune complexes (antibody/receptor) and complement activation.96 There is also a genetic susceptibility linked to an HLA-DQA1 allele located on 6p21,107 such that homozygous individuals have a predisposition for producing antibodies not only against PLA2R, but against other antigens as well.
An association has also been described between certain polymorphisms of PLA2R and the risk of MN, and in the same study, both protective and at-risk haplotypes were described for the disease, although these are not correlated with prognosis.108 The fact that the receptors of PLA2 are also found in other parts of the body, such as the lungs and leukocytes, indicates that there must be other local variables that explain why the clinical presentation is limited to renal involvement. A soluble form of PLA2R has been identified that is produced through alternative splicing and that supposedly has regulatory functions over the quantity of free circulating phospholipase A2. However, no studies have been able to demonstrate the existence of elevated levels of immune complexes that contain the soluble form of the receptor in MN patients,96 which has been interpreted as evidence in favour of the in situ formation of the immune complexes. The absence of antibodies against the soluble form of the receptor, whose molecular weight and structure are different from those of the membrane receptor, coincides with the evidence that the immunogenicity of the protein requires the preservation of certain antigenic conformations that are only present in the structure of the membrane protein.96 The discovery of IgG antibodies directed against PLA2R in a high percentage of patients with primary MN has allowed for the development of techniques for measuring circulating levels and specific stains for detecting its presence in renal biopsies. Both procedures constitute very relevant clinical advancements for the diagnosis and care of MN patients, with the potential to facilitate the identification of patients with primary forms of the disease, or to provide information regarding the activity of the disease at any point in time, thus orientating therapeutic decisions. Although the diagnostic value of this parameter appears unquestionable in light of the published results, further studies are needed in order to clearly define the prognostic value and potential usefulness of this molecule as an early indicator of recurrence.
The identification in humans of an antigen specific to the podocyte membrane against which IgG antibodies act in situ indicates that there are probably more local antigens implicated in these processes. Shortly after the identification of PLA2R as a target for the autoimmune response, two new auto-antibodies were discovered against podocyte antigens: aldose-reductase and manganese superoxide-dismutase 2 (SOD2), which colocalise with IgG deposits and complement and are selectively recognised by the IgG4 eluted from the renal parenchyma.109 These antibodies also appear to be specific to primary MN, since they have not been found in patients with secondary forms of MN or other renal diseases. The prevalence of these antibodies has not been determined in clinical studies, and until now, no clear aetiopathogenic link has been established. Ultrastructural analyses place the respective antigens in the cytoplasm of podocytes and podocyte processes, and the available evidence indicates that both antigens are neo-expressed by the podocytes, since in the healthy kidney, they are exclusively located in the tubules. In the case of SOD2, in vitro results associate its expression in the podocytes with oxidative stress. The fact that IgG4 recognises this molecule adds it to the list of new antigens identified as possible targets for the autoimmune response in primary MN. However, it is unknown what factor triggers the neo-expression of the antigen, nor what is the time-span of this pathway, such that we are currently unable to determine whether the autoimmune response observed is the primary trigger for the disease, or as occurs in other inflammatory processes, whether this is simply the consequence of a lesion in the podocyte caused by other immunological agents, which would trigger a secondary autoimmune response.
Antigens of extra-renal origin are responsible for the majority of cases of secondary MN. It is believed that these antigens trigger an immune response once they become deposited in the external membrane surface, transported through the circulatory system and passing through the glomerular basement membrane. These antigens can have multiple sources, primarily in association with systemic autoimmune, infectious, or neoplastic processes, or following exposure to certain drugs or food antigens. Recently, cases of MN have been described in which the renal lesion is produced as the result of the deposition of immunocomplexes formed by bovine albumin and anti-bovine albumin IgG.110
C4d staining in renal biopsies
Although it is not a circulating biomarker, in certain circumstances, the analysis of the complement activation pathway in renal biopsies can be useful for diagnosing nephrotic syndrome caused by MN. The presence of C4d in renal biopsies is considered to be evidence of complement activation through the classical pathway or the lectin pathway, but not the alternative pathway.111 The usefulness of C4d staining is widely recognised in the analysis of pathologies in kidney transplants, and more recently, its use has been expanded to the study of primary nephropathies. The existence of C4d deposits in biopsies from MN patients has been known for some time,112 but its potential clinical applicability had not been evaluated until only recently. In MN that appears in the native kidney, the histopathological diagnosis does not tend to be hindered by any doubts due to the characteristic results in the optical microscope, immunofluorescence, and electron microscope analyses. Recently, the possibility of using C4d staining using immunohistochemical techniques in paraffin-encased material has been described.113,114 In special cases for which insufficient material is available for immunofluorescence or electron microscopy, the evidence provided by a positive C4d stain in glomerular capillaries can be useful for distinguishing between MN, MCN, and FSG when optical microscope results are inconclusive. In a very recent study, it was shown that in MN patients that receive a kidney transplant, C4d positivity in glomerular capillaries in post-transplant biopsies may be a sign of recurrence of the primary nephropathy that precedes morphological changes that are characteristic of the disease, and as a consequence, could facilitate early diagnosis and treatment.115
Tubular proteinuria as a guide for indicating immunosuppressant treatment in membranous nephropathy
Criteria for indications and the moment for starting immunosuppressant treatment in patients with primary MN continues to be the subject of much debate due to the evidence that approximately 30%-40% of patients can develop spontaneous remission, whereas a similar percentage will progress towards renal failure if treatment is not provided.116-119 The clinical variables indicative of a poor prognosis are well defined, and spontaneous evolution in either direction tends to become evident within the first 2 or 3 years of follow-up. The clinical guidelines120 typically recommend a trial period using conservative treatment including angiotensin II receptor blockers before indicating immunosuppressant treatment. By medical consensus, it is generally recommended that this period last a minimum of 6 months, but the duration must be established on an individual basis for each case, based on the number of poor prognostic factors.
However, the evidence upon which these recommendations are based is not concrete enough to prevent some authors questioning and arguing that, even taking into consideration the observation periods recommended in the guidelines, there is a certain level of risk that many of these patients will be unnecessarily exposed to the toxicity of immunosuppressants.119 These groups suggest limiting immunosuppressant treatment to patients with the greatest risk of suffering progressive renal failure.116,121 Although it has been shown that this strategy can be used with high renal survival rates,121,122 the difficulty lies in coming to a consensus regarding which is the best parameter for identifying high-risk patients.
Those that defend restricting treatment initially considered that the evidence for deteriorated renal function was the most specific marker for indicating treatment.116,123,124 However, delaying the start of treatment until observing evidence of deteriorated renal function116,121 may favour the progression of the renal lesions towards a state of fibrosis, limit the efficacy of treatment, yield incomplete response to treatment, or even produce residual renal failure. In this context, medical researchers have searched for surrogate markers that facilitate predicting a prognosis during early stages of the disease and orientating a therapeutic decision, before renal function has deteriorated. The majority of the available information on this subject is centred on analysing the capacity of urine levels of tubule protein as a surrogate marker for the early detection of deteriorated renal function.125-129 Branten et al.126 analysed the validity and precision of urine excretion of beta-2 microglobulin (β2m) and IgG as predictors for the appearance of renal failure, defined as an increase in creatinine >50% or serum creatinine >1.5mg/dl, in a cohort of patients with MN and normal basal renal function. Patients received immunosuppressant treatment only when there was evidence of worsened renal function. A total of 44% of patients had deteriorated renal function, a value that coincides with the results from previous studies regarding the spontaneous evolution of untreated patients. In all cases, the deterioration was produced during the first 36 months of follow-up. Urine excretion of β2m≥0.5µg/min and IgG≥250mg/24h was associated with a risk of progression with the same sensitivity and with greater specificity and positive predictive value than proteinuria, for which the authors proposed that measuring both parameters could be helpful for deciding whether or not to start immunosuppressant therapy. However, the value of these data in clinical practice is very difficult to define, since no multivariate analyses were carried out to identify the independent predictors of disease progression, nor has the variability of urine levels of β2m and IgG been described when measured in the same patient over a long period of time. In addition, in accordance with the study results, basing a therapeutic decision on β2m or IgG levels would imply not treating 10% of patients, who would finally suffer renal function deterioration.
It has been suggested that urine levels of α1-microglobulin and other low-molecular weight proteins could have a similar predictive value,127 but none of these has been validated in independent clinical studies. Measuring β2m poses issues in daily clinical practice because urine can only be used if its pH is >6, since this molecule is degraded in acidic urine. As such, other tubule proteins with a more stable profile have been proposed. A good correlation has been described between urine excretion of N-acetyl-β-D-glucosaminidase (NAG), whose levels do not depend on urine pH, and the prognosis of MN patients.128 In a recent study129 that compared the predictive value of β2m with NAG, the authors concluded that, although both can be useful for predicting prognoses, β2m is the more precise of the two. In this study, the multivariate analysis yielded urine excretion of β2m as the best independent predictor of deterioration of renal function.
Altogether, although no external validation has been produced, these results suggest that urine levels of β2m and NAG could be useful for monitoring patients during the observation phase prior to making a decision regarding treatment. The presence of elevated β2m or NAG levels would be associated with a poor prognosis and could be a variable to take into account when making decisions regarding immunosuppressant treatment. However, the absence of information regarding the variability of β2m and NAG levels, when measured in the same individual over long periods of time, along with the evidence that approximately 15% of patients with low levels of β2m have deteriorated renal function and the fact that almost 20% of patients with high β2m levels can enter into spontaneous remission, hinders defining the true clinical usefulness of these parameters. On the other hand, considering the basal glomerular filtration rate in these patients (71±23ml/min/1.73m2), it is obvious that this study included patients with reduced basal renal function.
This fact impedes evaluating the capacity of β2m and NAG for predicting the evolution of renal function before it deteriorates, since in many cases the deterioration has already taken place, and also impedes extrapolating the results of this study for use in patients with normal renal function. Additionally, the inclusion of patients with reduced basal renal function can be very important when interpreting the described independent predictors of the evolution of renal function, since both β2m and NAG can be indicators of more advanced renal lesions, as these molecules reflect the extent of tubulo-interstitial damage.130 As such, it is probable that patients with higher levels of β2m and NAG also had lower initial glomerular filtration rates, and as a result, a lower probability of spontaneous remission or response to treatment.
Conflicts of interest
The authors have no conflicts of interest to declare.
Key concepts
1. Measuring urine levels and/or podocyte expression of CD80 may be useful for the non-invasive diagnosis of MCN as well as for the differential diagnosis between MCN and FSG.
2. In MCN patients, during the breakout phase, a circulating isoform of Hx has been identified with increased protease activity, although the clinical significance of this finding is poorly understood.
3. The circulating levels of sIL-2R are elevated during the breakout phase of MCN, but more studies are needed to analyse whether measuring these levels provides any added value in the diagnosis or prognosis of MCN as compared to conventional tests.
4. Quantifying the expression of ABCB1-glycoprotein-P in lymphocytes could be useful for predicting the response to steroid treatment, the risk of later recurrences, and/or for suggesting the early indication of other treatment options.
5. Determining the clinical value of suPAR as a possible FSG biomarker requires further studies in order to determine what levels have a diagnostic value and whether the presence of elevated levels of suPAR has any correlation with the clinical presentation, response to treatment, or prognosis of the disease.
6. The presence of elevated levels of anti-PLA2R antibodies has a high specificity for the diagnosis of primary MN, these levels are correlated with the clinical activity of the disease, and, in transplanted patients, this measurement can be useful for differentiating between a recurrence of MN and de novo MN.
7. Currently, the clinical significance of circulating auto-antibodies against aldose-reductase and manganese superoxide-dismutase 2 in MN is unknown.
8. In MN patients with normal renal function, elevated β2m and NAG levels in urine samples as measured during the initial period following diagnosis could be useful for making the decision to start immunosuppressant treatment.

Table 1. List of characteristics and potential usefulness of the primary biological markers proposed for the study of nephrotic syndrome

Table 2. Summary of the clinical study results and types of auto-antibodies described for the study of membranous nephropathy

Figure 1. Diagram of the pathogenic mechanisms proposed to explain the podocyte damage in minimal change nephropathy and focal segmental glomerulonephritis based on the possible biomarkers found in the study phase



