



Dear Editor,
Secondary systemic amyloidosis (AA) is a frequent condition, associated with long evolution inflammatory and infectious diseases, as also with some neoplasias. During the first 15-20 years, the disease presents no symptoms, and usually renal involvement is the first clinical sign to appear and manifests itself in the form of proteinuria. It usually evolves to terminal renal failure 2-10 years later.1 Furthermore, amyloidosis relapse during kidney transplant is also a widely described condition.2 Primary (AL) amyloidosis or de novo secondary (AA) amyloidosis during renal transplant, are considered slowly progressive diseases, which in most cases are not associated with graft loss3 or only cause graft loss after years of evolution.4
Gangrenous pyoderma (GP) is a well-defined skin clinical pathological condition, characterised by the presence of single or multiple erythematous pustules that rapidly progress to necrotic ulcers. Its cause is unknown, although some defects of the oxygen metabolism of neutrophils, overexpression of cytoquines (interleukin-8, interleukin-16) and anomalies of humoural and cell immunity have been described as possible causes, but none is specific.5 In mild forms, treatment with topical or intralesional steroids is used, but it is always insufficient, and systemic treatment is necessary. Cases that are refractory to steroids can respond to other immunosuppressants, such as oral cyclosporin and sometimes mofetil mycophenolate, azathioprine or methotrexate are also effective, as also new biological therapies (anti-TNF monoclonal antibodies).
We present the case of a 38 year old woman, with a history of well controlled hypertension of long evolution and GP of the lower limbs diagnosed in 1989. Her GP was treated with calcineurin inhibitors (cycosporin and tacrolimus), without any response. She presented multiple infectious complications due to multi-resistant germs that made prolonged antibiotic therapy necessary. Monitored in our service for chronic kidney disease (CKD), a renal biopsy was performed in February 2002, and the report informed of the presence of sclerosis and patchy glomerular atrophy, with vascular hyalinosis, compatible with anticalcineurin treatment side effects.
In August 2002, the patient began substitute renal therapy with haemodialysis, and in October 2005 received a kidney transplant from a deceased donor. On discharge, plasma creatinine levels were 1 mg/dl without proteinuria. For 4 years she received therapy with tacrolimus, mofetil mycophenolate and prednisone at low doses. Renal function was stable, with plasma creatinine of around 1.1 mg/dl and proteinuria kept at <0.5 g/24 hours in spite of presenting several complications in the form of secondary infections to GP. In February 2009 renal function deteriorated (plasma creatinine 1.5 mg/dl) and there was also an increase in proteinuria (2.8 g/24 hours) (Table 1). The possible differential diagnoses were: chronic graft nephropathy, toxicity due to calcineurin drugs, de novo glomerular disease, tubule-interstitial or obstructive nephritis. On physical exam the patient was normotense and presented decrease of adipose tissue related to long term protein-calorie malnutrition, lesions in lower limbs secondary to GP and mild discomfort on palpation of the renal graft area. A complete laboratory profile was obtained for glomerular diseases, including a proteinogram and electroimmunophoresis in blood and urine; all tests were negative.
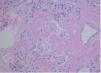
On kidney ultrasound a graft measuring 13.2 x 6.2 cm was detected, with preserved parenchyma and echostructure with no obstructive evidence. On 23rd May 2009 a renal biopsy was performed and 25 glomeruli were obtained in which a variable amount of eosinophil amorphous material was observed. In most glomeruli it was deposited in the glomerular tuft and extended to the remainder of the glomerulus. Such material was also seen in the interstitium, surrounding the tubules and in the vessel wall thickness. It was even more clearly seen when apple green bright Congo Red stain was used under polarised light (Figure 1). When pretreated with potassium permanganate, the Congo Red stain was lost. It was also possible to see tubules with edematous epithelium and a slightly marked small-sized lymphocyte chronic inflammatory infiltrate in the interstitium. The immunofluorescence study was negative. The anatomopathological diagnosis was type AA secondary amyloidosis. By means of ultrasound studies, infiltrating cardiomyopathy was ruled out, as also infiltrations in other locations. On the 25th of June, 3 months after the beginning of the clinical condition, the patient began substitute renal therapy, and died 6 months later due to cerebral haemorrhage with low platelets and graft intolerance.
We present the case of a 36 year old woman, with AA de novo amyloidosis in the kidney graft in the context of a chronic inflammatory process; these facts are widely described in the literature, as mentioned before, but this case has several singular characteristics that led us to present the condition, so that it may serve to reflect on when in the presence of other cases of acute kidney graft deterioration. In the first case, we present the first case described in the literature of an association of GP, a chronic inflammatory process, as we have mentioned before, with AA amyloidosis. Secondly, the appearance of de novo AA amyloidosis in a kidney graft, without systemic involvement, after 16 long years of inflammatory and infectious processes associated with lower limb ulcers, the manifestation of the patient's GP. Cases have been reported in the scientific literature in which systemic amyloidosis develops after a long inflammatory process, or even does so in a kidney graft, but after years of inflammation. In this case, after 16 years of inflammation the patient did not develop amyloidosis but did do so in only 4 years after kidney transplant with the amyloidosis developing in this organ. In the third place, both AL and AA de novo amyloidosis, according to what has been described in the medical literature so far, cause a slow and insidious deterioration of renal function. In our patient, loss of graft occurred in only 3 months, with subsequent death.
This case should be something to reflect on when establishing a differential diagnosis in patients with kidney graft and associated chronic inflammatory processes who begin to suffer rapid deterioration of their function associated with proteinuria and in which all laboratory and clinical results are negative.

Table 1. Evolution of plasma creatinine and proteinuria

Figure 1. Amyloidosis in renal biopsy glomerulus



