
Bartter syndrome (BS) is a rare disease with an incidence of approximately 1/1,000,000 population. Currently, BS is classified into 5 types according to the genetic variant identified (Table 1).1–4 There are few studies describing the long-term evolution of these patients.
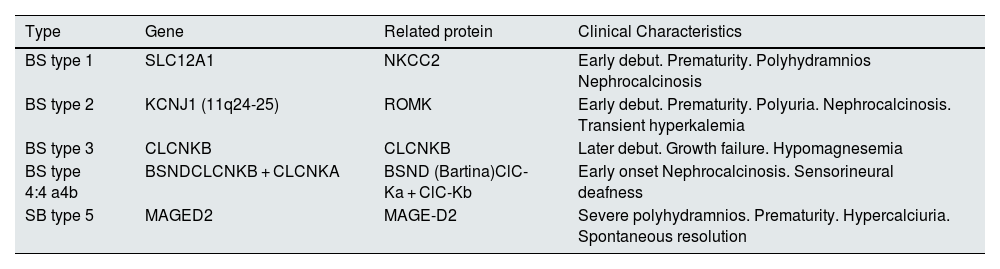
Types of Bartter syndrome.
| Type | Gene | Related protein | Clinical Characteristics |
|---|---|---|---|
| BS type 1 | SLC12A1 | NKCC2 | Early debut. Prematurity. Polyhydramnios Nephrocalcinosis |
| BS type 2 | KCNJ1 (11q24-25) | ROMK | Early debut. Prematurity. Polyuria. Nephrocalcinosis. Transient hyperkalemia |
| BS type 3 | CLCNKB | CLCNKB | Later debut. Growth failure. Hypomagnesemia |
| BS type 4:4 a4b | BSNDCLCNKB + CLCNKA | BSND (Bartina)ClC-Ka + ClC-Kb | Early onset Nephrocalcinosis. Sensorineural deafness |
| SB type 5 | MAGED2 | MAGE-D2 | Severe polyhydramnios. Prematurity. Hypercalciuria. Spontaneous resolution |
We conducted a retrospective study including 19 cases with the clinical diagnosis of BS (from 1969 to 2021), 10 with genetic confirmation: 5 mutation in KCNJ1 and 5 in CLCNKB. The median age at the last visit was 17 years (IQR: 6.93).
Twelve were diagnosed in the first year of life and 7 between 1 and 4 years of age (5 born before 1975). Polyhydramnios was recorded in 12 cases, and 9 were preterm.
The reason for consultation was stagnation of weight and height and/or gastrointestinal symptoms in 12, hydroelectrolyte disturbances in 5 and the finding of nephrocalcinosis in 2.
All received continuous treatment with nonsteroidal anti-inflammatory drugs (NSAIDs), indomethacin (maximum dose 2.04 ± 0.68 mg/kg/day) or Tolmetin (maximum dose 31.2 ± 14 mg/kg/day), with no significant adverse effects that required discontinuation. The 2021 guidelines recommend its use with caution and with possible interruptions at school age to avoid a prolonged use.3 They suggest monitoring renin levels to reduce the dose of NSAIDs until renin levels are within the normal range.5 We have not performed this monitoring and have maintained the minimum dose necessary to achieve good clinical and metabolic control. In addition, they received prophylactic proton pump inhibitors.
Oral potassium supplementation was variable, with a maximum dose between 0.5 and 15 mEq/kg/day (median: 4.4). Magnesium supplementation was required in 36% of patients.
The mean initial height was −2,1 ± 1,65 SD, median −1,58 (IQR: 2,77). The evolution of height was satisfactory with a mean increase of +1.07 ± 1.08 SD. Several factors have been implicated in growth retardation: on the one hand, metabolic disturbances (alkalosis, chronic hypokalemia, salt depletion), and also the possibility of partial or complete growth hormone (GH) deficiency.3,6 In our study only a single case with BS type3 has a height below 2 SD at the last visit ; this patient being still under study and initiating puberty, although she has improved height SD since diagnosis and with very good therapeutic compliance, she was the patient with lowest K+ values, below 3 mEq/l, and GH deficit was ruled out.
In the initial study 13 patients had an elevated urinary Ca/Cr index for their age and 11 (58%) had nephrocalcinosis by ultrasound, a higher incidence than that reported in the literature, which varies between 25%7 and 53.8%.8 However, only 20% of the BS type 3 presented nephrocalcinosis, similar to that described in other series.9 In the last control, 3 patients maintained an elevated urinary Ca/Cr ratio.
Patients with BS have multiple factors for developing chronic kidney disease (CKD): being premature, low birth weight, episodes of severe dehydration, nephrocalcinosis, proteinuria secondary to hyperfiltration and treatment with NSAIDs.1,2 The incidence in the different series is very variable, many are not homogeneous in terms of age because some include adult patients and because of the difference in genetics, since it is know that CKD is much more frequent in BS type1 and type 4.1,8,10 In our series only 3 patients presented CKD stage II, all of them were BS type 2, with severe nephrocalcinosis and major prematurity.
One of the important elements in the follow-up of these patients is the be aware of the warning signs indicating the need to attend the emergency department. These patients are instructed to go to the emergency department especially if they present vomiting with poor oral tolerance; in these cases, serum therapy for a few hours can avoid admission. Although we have not found published data on the number of admissions after diagnosis of these patients, in our study 63% required hospitalization, half of them only one admission and most of them due gastrointestinal symptoms. Only 2 cases had multiple admissions for decompensation or medication adjustments due to poor adherence to therapy.
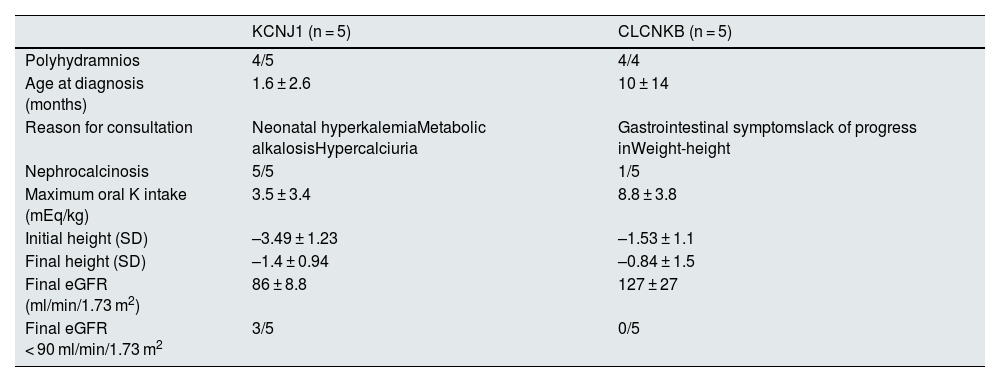
In the 10 patients for whom genetic studies were available, 5 had a mutation in KCNJ1 and 5 had a mutation in CLCNKB. The most significant findings in these cases are described in Table 2.
Most relevant findings in the 10 patients with genetic study.
| KCNJ1 (n = 5) | CLCNKB (n = 5) | |
|---|---|---|
| Polyhydramnios | 4/5 | 4/4 |
| Age at diagnosis (months) | 1.6 ± 2.6 | 10 ± 14 |
| Reason for consultation | Neonatal hyperkalemiaMetabolic alkalosisHypercalciuria | Gastrointestinal symptomslack of progress inWeight-height |
| Nephrocalcinosis | 5/5 | 1/5 |
| Maximum oral K intake (mEq/kg) | 3.5 ± 3.4 | 8.8 ± 3.8 |
| Initial height (SD) | –3.49 ± 1.23 | –1.53 ± 1.1 |
| Final height (SD) | –1.4 ± 0.94 | –0.84 ± 1.5 |
| Final eGFR (ml/min/1.73 m2) | 86 ± 8.8 | 127 ± 27 |
| Final eGFR < 90 ml/min/1.73 m2 | 3/5 | 0/5 |
The fundamental limitation of our study is that we have only collected the patients followed up in our unit, so we do not have the genetic study in 9 of the cases, all of them transferred to adult units before 1998. Unlike other published studies, which describe a prevalence of BS type 1 of 22.8%, we only had 10 patients with a genetic study that had mutations compatible with BS type 2 and type 3.



